


2. 西北大学化工学院/陕西省物理无机化学重点实验室, 陕西 西安 710069;
3. 故宫博物院文保科技部, 北京 100009
2. College of Chemical Engineering/Shaanxi Key Laboratory of Physico-Inorganic Chemistry, Northwest University, Xi′an 710069, China;
3. Conservation Technology Department, The Palace Museum, Beijing 100009, China
四嗪、四唑类化合物,是继呋咱化合物后近几年国外研究较多的一类新型高氮化合物,因其分子结构中含有大量的N—N和C—N键,故其普遍具有很高的正标准生成焓,有别于传统的含能化合物,其化学潜能主要来源于其正标准生成焓;同时,因其分子结构中碳、氢含量低,使其出现双重正效应:既能提高材料的密度,又容易调节氧平衡[1]。这些高氮化合物作为推进剂组分,可以调节燃烧产物的平均分子量,有利于提高比冲;同时又可以减少推进剂的烟雾,是一类新型高氮含能材料。
3, 6-双(1-氢-1, 2, 3, 4-四唑-5-氨基)-1, 2, 4, 5-四嗪(BTATz)是美国Los Alamos国家实验室率先设计合成的一种新型高氮含能化合物,氮含量达80%,生成焓为+883 kJ·mol-1;其DSC起始分解点264 ℃,有较好的热稳定性[2-3];燃烧性能方面,BTATz表现出高燃速、低燃速压强指数的特点[4];此外,BTATz在固体推进剂中的应用有利于降低推进剂的特征信号[5],在燃气发生剂、新型无烟灭火剂和富燃料推进剂中也有应用潜力[6-7]。目前,国内外科学家已对BTATz的合成、燃烧性能[8-12]进行了初步探讨,但其量子化学研究报道很少。
分子间相互作用研究可预示体系分子的结合力并预测其稳定性,因而对材料的配方设计有着重要的意义。并且由于爆炸物的许多物理、化学和爆炸性质均与其聚集状态有关,故须借助分子间相互作用研究加以阐明。本文对BTATz二聚体进行了密度泛函理论计算,揭示了BTATz二聚体的稳定几何构型、电子结构、及其结合能和稳定性的关系,为深入研究该类化合物的结构-性能关系提供了基础数据。
2 计算方法在DFT-B3LYP/6-31G*水平下,先对BTATz孤立分子进行几何构型全优化和电子结构计算;然后利用Chem3D软件组建得到(BTATz)2势能面上的可能稳定构型并用Berny方法[13]进行全优化。分子间相互作用能的计算采用Boys和Bernardi[14]提出的均衡校正方法(CP)以消除基组重叠误差(BSSE),其表达式为:
| $ \begin{array}{l} \Delta {E_{\rm{c}}} = {E_{{{\rm{A}}^*}{{\rm{B}}^*}}} - {E_{\rm{A}}} - {E_{\rm{B}}} + {\rm{BSSE}}\\ = {E_{{{\rm{A}}^*}{{\rm{B}}^*}}} - {E_{\rm{A}}} - {E_{\rm{B}}} + {E_{{{\rm{A}}^*}}} + {E_{{B^*}}} - {E_{{{\rm{A}}^*}({{\rm{A}}^{\rm{*}}}{{\rm{B}}^*})}} - {E_{{{\rm{B}}^*}({{\rm{A}}^*}{{\rm{B}}^*})}} \end{array} $ |
式中,A*B*为二聚体的优化构型;EA*B*为二聚体A*B*的能量;EA为单体A的能量;EA*为子体系A*取二聚体构型时的单点能;EA*(A*B*)则为选用二聚体A*B*的所有基集时EA*的单点能;子体系B的各种能量含义与A相同。
通过自然键轨道分析揭示电荷转移状况和分子间相互作用的本质。基于统计热力学方法求得热力学函数。全部计算均采用Gaussian 98[15]程序在lenovo P4微机上完成,收敛精度取程序内定值。
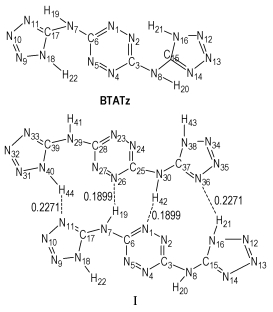
3 结果与讨论 3.1 几何构型BTATz单体及其二聚体的6种稳定结构如图 1所示,在DFT-B3LYP/6-31G*水平下BTATz单体及其6种二聚体的部分全优化几何参数列于表 1。BTATz单体,二聚体Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ均属C1点群。由表 1可见,与单体分子键长相比,二聚体的键长变化均主要发生在两分子间氢键所形成的环上。例如,二聚体Ⅰ的键长变化主要发生在氢键N(40)—H(44)…N(11)和N(7)—H(19)…N(26)以及氢键N(30)—H(42)…N(1)和N(16)—H(21)…N(36)所分别形成的十一元环上,而其余键的键长变化相对较小。在二聚体Ⅰ中,N(7)—H(19)、N(30)—H(42)增加1.6 pm,C(3)—N(4)、N(11)—C(17)、C(28)—N(23)和N(36)—C(37)增加0.6 pm,N(8)—C(15)、N(29)—C(39)增加0.8 pm,N(40)—H(44)、N(16)—H(21)增加1.1 pm,而N(7)—C(17)、N(30)—C(37)减小0.6 pm,C(28)—N(29)、C(3)—N(8)减小0.9 pm。在二聚体Ⅱ中N(7)—H(19)、N(30)—H(42)增加2.9 pm,N(11)—C(17)、N(36)—C(37)增加0.9 pm,而N(7)—C(17)、N(30)—C(37)减小1.1 pm。其余二聚体分别与Ⅰ、Ⅱ结构相似且键长变化也与其基本一致。6种二聚体的键角变化均在5.4°之内。二面角变化为:二聚体Ⅰ中,D(6, 7, 17, 11)、D(6, 7, 17, 18)、D(25, 30, 37, 36)和D(25, 30, 37, 38)分别增加12.86°、13.39°、12.84°和13.37°,而D(1, 6, 7, 17)、D(5, 6, 7, 17)、D(3, 8, 15, 16)、D(24, 25, 30, 37)、D(26, 25, 30, 37)和D(28, 29, 39, 40)分别减小9.93°、10.58°、5.61°、10.58°、9.91°和5.60°。说明分子间相互作用使单体中—NH—发生了旋转,并且参与分子间氢键的—NH—旋转相对更明显。其余二聚体均各子分子的平面构型均没有发生变化,即二面角几乎无变化,故表中未予列出。

|
图 1 BTATz及其二聚体的优化构型、分子间距离(nm)和原子编号 Fig.1 Optimized geometries and atomic numbering of BTATz and its dimers, and intermolecular distance(nm) |
| 表 1 DFT-B3LYP/6-31G*水平下BTATz和(BTATz)2的部分全优化几何参数 Tab.1 The optimized geometry parameters of BTATz and (BTATz)2 at DFT-B3LYP/6-31G* level |
由图 1可见,二聚体Ⅰ、Ⅲ、Ⅳ均存在四个氢键N—H…N,二聚体Ⅱ、Ⅴ、Ⅵ均存在两个氢键N—H…N。按通常观点,当接触点数相同时,分子间结合能(分子间作用能的负值)大小由分子间距离长短所决定,这里自然应由分子间的氢键强弱所决定,即可预示二聚体的结合能和稳定性排序为:Ⅲ>Ⅳ>Ⅰ;Ⅱ>Ⅵ>Ⅴ。
3.2 相互作用能表 2列出在DFT-B3LYP全优化构型下BTATz及其二聚体的零点校正能(ZPEC)、基组叠加误差(BSSE)和经BSSE和ZPEC校正前后的分子间相互作用能。由表 2可见,构型Ⅱ经校正后结合能(最大)为-68.82 kJ·mol-1,每个氢键平均提供结合能为-34.41 kJ·mol-1,可见该氢键非常强!在B3LYP/6-31G*水平下,比较ΔE和(ΔE)C,ZPEC可知,由B3LYP法计算(BTATz)2之间的结合能必须进行BSSE和ZPEC校正,这不仅极大地影响结合能的数值,而且导致各构型结合能排序出现交错。如未校正的相互作用能|ΔE|的大小顺序为:Ⅱ>Ⅲ>Ⅳ>Ⅰ>Ⅵ>Ⅴ,经BSSE和ZPEC校正后的相互作用能|(ΔE)C,ZPEC|的大小排序为:Ⅱ>Ⅲ>Ⅳ>Ⅵ>Ⅰ>Ⅴ。这与前面根据氢键长短来推测的聚合体稳定性相一致,表明氢键在分子间相互作用中起重要作用。
| 表 2 DFT-B3LYP水平下的零点能和相互作用能 Tab.2 Zero point energy and binding energies at the DFT-B3LYP level |
为了确定基组的适合性,对B3LYP/6-31G*水平下所得优化构型用6-311++G**基组作单点能计算。比较该2种基组下所得结果可知,不同基组时所有二聚体的BSSE校正结合能基本一致。而且经BSSE校正后的相互作用能|(ΔE)C|的大小排序与在B3LYP/6-31G*水平下的|(ΔE)C,ZPEC|排序完全一致。
3.3 电荷分布和转移表 3列出各原子上净电荷。与单体相比,二聚体Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ的电荷变化主要发生在形成氢键附近的C、N、H原子上。如在二聚体Ⅰ中,N(2)、N(27)、C(6)、C(25)、N(10)、N(35)、N(12)、N(31)、C(17)、C(37)、H(21)和H(44)原子分别增加0.0243 e、0.0243 e、0.0156 e、0.0156 e、0.0148 e、0.0148 e、0.0117 e、0.0117 e、0.0210 e、0.0210 e、0.0103 e和0.0103 e的电荷,而N(1)、N(26)、N(7)、N(30)、N(11)、N(36)、C(15)和C(39)原子分别减小0.0594 e、0.0594 e、0.0162 e、0.0162 e、0.0440 e、0.0440 e、0.0121 e和0.0121 e的电荷。二聚体Ⅲ中,N(2)、C(6)、N(10)、C(17)、N(26)、C(28)、N(33)、N(35)、N(38)和H(41)原子增加0.0108~0.0283 e的电荷,而N(1)、N(7)、N(11)、C(15)、N(23)和N(34)原子减小0.0107~0.0602 e的电荷。其余二聚体的电荷变化与二聚体Ⅰ、Ⅱ的类似,故不再赘述。从而表明所有二聚体的电荷变化比较大。然而各构型子体系之间净电荷转移几乎为零。BTATz单体及二聚体Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ和Ⅵ的偶极矩分别为0.0043、1.1797、0.0035、0.6323、0.0046、0.0012、0.1942 Debye。
| 表 3 DFT-B3LYP/3-21G*水平下(BTATz)2的自然原子电荷(e) Tab.3 The calculated natural atomic charges (e) of (BTATz)2 at the DFT-B3LYP/3-21G* level |
NBO电子供体(Donor)轨道i、电子受体(Acceptor)轨道j和它们之间相互作用的稳定化能E列于表 4。NBO轨道相互作用强度与稳定化能E成正比。
| 表 4 (BTATz)2的B3LYP/3-21G*自然键轨道分析部分结果 Tab.4 Parts of calculated results of (BTATz)2 at the B3LYP/3-21G* level by NBO analysis |
由表 4可见,氢键最强的构型Ⅱ中N(11)的孤对电子(1)对N(30)—H(42)的σ反键轨道的稳定化能为125.44 kJ·mol-1,N(36)的孤对电子(1)对N(7)—H(19)的σ反键轨道的稳定化能为125.14 kJ·mol-1,说明构型Ⅱ主要是通过N(11)和N(36)的孤对电子分别和N(30)—H(42)以及N(7)—H(19)的σ反键轨道之间相互作用的;构型Ⅲ中N(1)的孤对电子(1)对N(38)—H(43)的σ反键轨道的稳定化能为49.87 kJ·mol-1,N(11)的孤对电子(1)对N(29)—H(41)的σ反键轨道的稳定化能为60.79 kJ·mol-1,N(23)的孤对电子(1)对N(7)—H(19)的σ反键轨道的稳定化能为119.37 kJ·mol-1,N(34)的孤对电子(1)对N(16)—H(21)的σ反键轨道的稳定化能为49.16 kJ·mol-1。说明构型Ⅲ的分子间作用主要发生在N(1)、N(11)、N(23)以及N(34)的孤对电子分别和N(38)—H(43)、N(29)—H(41)、N(7)—H(19)和N(16)—H(21)的σ反键轨道之间。
类似分析表明,构型Ⅰ、Ⅳ、Ⅴ、Ⅵ的分子间作用也均主要发生在N的孤对电子与邻近的N—H的反键轨道之间。均证实氢键是主要作用并揭示了氢键作用的本质。
3.5 热力学性质基于统计热力学方法,在振动分析基础上,用B3LYP频率(校正因子为0.96)分别计算了BTATz和(BTATz)2在200.0~800.0 K的标准恒压热容(Cpθ)、标准熵(Smθ)和标准焓(Hmθ)。进而计算由单体形成二聚体的热力学性质变化(ΔST,ΔHT和ΔGT)。结果一并列于表 5。
| 表 5 不同温度下BTATz和(BTATz)2的热力学性质 Tab.5 The thermodynamic properties of BTATz and (BTATz)2 at different temperatures |
不难发现,在同一温度下6种二聚体的Cpθ值基本接近,同时,分子间相互作用使它们比BTATz单体的2Cpθ大7.10~15.31 J·mol-1 K-1。由单体形成二聚体的有序度增加,故熵值减少。分子间相互作用是一个放热过程,由单体形成二聚体,体系焓值减少;在同一温度下,焓变的次序为:(ΔHT)Ⅴ>(ΔHT)Ⅵ>(ΔHT)Ⅰ>(ΔHT)Ⅳ>(ΔHT)Ⅲ>(ΔHT)Ⅱ,结合能顺序为:Ⅱ>Ⅲ>Ⅳ>Ⅰ>Ⅵ>Ⅴ(与未经校正的结合能顺序一致)。由ΔGT=ΔHT-TΔST求得不同温度下的ΔGT值。发现在200 K时所有ΔGT均为负值,表明低温下单体形成二聚体均能自发进行;在常温下,除二聚体Ⅴ外,其它二聚体也均能自发形成。在400 K由单体能自发进行形成二聚体Ⅱ、Ⅲ。在所研究过的各高能体系中,还没有出现过在400 K下二聚过程能自发进行的例子。比较各温度下ΔGT的相对大小,发现在400 K之下,二聚体的稳定性排序为Ⅱ>Ⅲ>Ⅳ>Ⅵ>Ⅰ>Ⅴ,表明ΔGT由ΔHT(即焓变或能量因素)所决定;而当温度在600 K以上时,ΔST成为决定ΔGT的主导因素,亦即体系规整度的影响增大,致使ΔGT亦即体系稳定性排序出现较复杂的变化。
4 结论(1) 在DFT-B3LYP/6-31G*水平上,求得的6种(BTATz)2二聚体的全优化几何构型。其中聚合体的最大相互作用能为-68.82 kJ·mol-1,属于构型Ⅱ。
(2) 分子间相互作用使得构型Ⅰ中—NH—发生了扭曲,而且参与氢键的—NH—扭曲得相对明显一些。各构型子体系之间几乎均无净电荷转移。
(3) 由单体形成二聚体是放热和熵减小过程。随温度升高,焓变和熵变的减小值均下降而吉布斯自由能的变化值却升高。在较低温度下由单体形成6种二聚体均能自发进行,而在常温下,除了二聚体Ⅴ外,其余二聚体亦均能自发形成。二聚体Ⅱ、Ⅲ甚至在400 K时能自发形成。
| [1] |
Hiskey M A, Chavez D E, Naud D. Progress in high-nitrogen chemistry in explosives, propellants and pyrotechnics[C]//Proceedings of 27th International Pyrotechnics Seminar. July 16-21, USA, Colorado, 2000: 3-14.
|
| [2] |
Chavez D E, Hiskey M A. 1, 2, 4, 5-Tetrazine based energetic materials[J].
Journal of Energetic Materials, 1999, 17: 357-377. DOI:10.1080/07370659908201796 |
| [3] |
Hiskey M A, Chavez D E. Insensitive high-nitrogen compounds. DE: 776133[R]. 2001.
|
| [4] |
Hiskey M A, Chavez D E. Propellant containing 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine or salts thereof: US 6458227[P]. 2002.
|
| [5] |
Hiskey M A, Chavez D E, Naud D. 3, 6-Bis (1H-1, 2, 3, 4-tetrazol-5-ylamino)-1, 2, 4, 5-tetrazine or salt thereof: US 6657059[P]. 2003.
|
| [6] |
鲍桐, 张炜, 阳世清, 等. BTATz在AP/DHG/HTPB型燃气发生剂中的应用研究[C]//中国宇航学会固体火箭推进专业委员会第二十一届年会固体火箭推进技术学术会议论文集. 上海. 2004.
|
| [7] |
Lu Y C, Wierenga P H. Advanced propellant/additive development for fire suppressing gas generators[C]//Proceedings of Halon Options Technical Working Conference. Sheraton Old Town Albuquerque. New Mexico. 2000.
|
| [8] |
岳守体, 阳世清. 3, 6-双(1-氢-1, 2, 3, 4-四唑-5-氨基)-1, 2, 4, 5-四嗪的合成及其性能[J].
含能材料, 2004, 12(3): 155-157. YUE Shou-ti, YANG Shi-qing. Synthesis and properties of 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12(3): 155-157. |
| [9] |
岳守体, 阳世清. 3, 6-双(1-氢-1, 2, 3, 4-四唑-5-氨基)-1, 2, 4, 5-四嗪的合成与表征[J].
合成化学, 2004, 12(2): 164-167. YUE Shou-ti, YANG Shi-qing. Synthesis and characterization of 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine[J]. Chinese Journal of Synthetic Chemistry, 2004, 12(2): 164-167. |
| [10] |
王伯周, 来蔚鹏, 刘愆, 等. 3, 6-双(1-氢-1, 2, 3, 4-四唑-5-氨基)-1, 2, 4, 5-四嗪的合成、表征及量子化学研究[J].
有机化学, 2008, 28(3): 422-427. WANG Bo-zhou, LAI Wei-peng, LIU Qian, et al. Synthesis, characterization and quantum chemistry study on 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine[J]. Chinese Journal of Organic Chemistry, 2008, 28(3): 422-427. |
| [11] |
Saikia A, Sivabalan R, Polke B G, et al. Synthesis and characterization of 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine (BTATz): Novel high-nitrogen content insensitive high energy material[J].
Journal of Hazardous Materials, 2009, 170: 306-313. DOI:10.1016/j.jhazmat.2009.04.095 |
| [12] |
张兴高, 朱慧, 阳世清, 等. 富氮高能物质BTATz的热分解动力学和分解机理[J].
推进技术, 2007, 28(3): 322-326. ZHANG Xing-gao, ZHU Hui, YANG Shi-qing, et al. Study on thermal decomposition kinetics and mechanism of nitrogen-rich compound BTATz[J]. Journal of Propulsion Technology, 2007, 28(3): 322-326. |
| [13] |
Baker J. An algorithm for geometry optimization without analytical gradients[J].
J Comput Chem, 1987(8): 563-574. |
| [14] |
Boys S F, Bernadi F. The calculation of small molecular interactions by the differences of separate total energies: Some procedures with reduced errors[J].
Mol Phys, 1970, 19(4): 553-566. DOI:10.1080/00268977000101561 |
| [15] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussisn 98, Revision A. 7, Pittsburgh PA: Gaussian, Inc., 1998.
|
Six optimized stable 3, 6-bis(1H-1, 2, 3, 4-tetrazol-5-yl-amino)-1, 2, 4, 5-tetrazine (BTATz) dimers were found on the potential energy surface and their electronic structures have been obtained by using density functional theory (DFT) at the B3LYP/6-31G* level, and the intermolecular interaction energy was calculated with basis set superposition error correction (BSSE) and zero point energy (ZPE) correction. The natural bond orbital (NBO) analysis was performed to reveal the origin of the interaction.