



2. Institute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621900, China
2. 中国工程物理研究院化工材料研究所, 四川 绵阳 621900
Octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine (HMX, C4H8N8O8) is one of the most widely used ingredients in various propellants and in several high-performance plastic-bonded explosives. HMX co-crystallizes with nearly one hundred organic molecules to produce stoichiometric solids. The firstly reported crystal structure of an HMX solvate is the 1: 1 complex between HMX and N, N-dimethylformamide (DMF, C3H7NO)[1-3]. Co-crystallization with other conformers provide a distinct approach to tailor physicochemical attributes, such as solubility[4], stability[5], bioavailability[6], hygroscopicity[7] and mechanical properties[8] of an active pharmaceutical ingredient (API) whilst maintaining the biological function of the drug. For the energetic materials, co-crystallization with other compounds also provides an opportunity to modify both the chemical composition and physical properties of the explosives through deliberate modification of the conformer. HMX as an ingredient in the vulnerability propellant formulation sometimes results in the reduction of the combustion velocity[9]. Co-crystallization with other compounds may be an approach to avoid this problem[10-11]. Besides, it can improve the sensitive and mechanical properties of HMX[12-13].
HMX is easy to co-crystallize with DMF other than re-crystallization in DMF solution. In our experiment of preparing cocrystal of HMX and TATB in DMF solution, by chance, we have obtained the single crystal of HMX/DMF solvate. HMX/DMF crystallizes in three different polymorphic forms, belonging to R3c, C2/c, and R3 space groups[1-3], respectively. Hereinto, R3c is the most stable crystalline form. In the polymorphic forms of HMX/DMF, all HMX molecules present α-conformation possessing a crystallographic 2-fold axis. In the R3c and C2/c crystal structures, the DMF molecule exhibits twofold disorder and no positions have been retained[1-2]. Experiment has also not given the atom coordinates for the R3 polymorph. So, the crystal structures of the HMX/DMF polymorphs have not been completely determined in experiments. Theoretically, the prediction of crystal structures of multi-component molecular crystals, including solvates, salts, and cocrystals, represents a major challenge in the field of crystal structure. So far, only several studies can be found[14-16]. Since the first reports of the crystal structures of HMX/DMF, fewer studies are devoted to mechanistic understanding of the co-crystallization process. In general, for a crystalline system, if homomeric intermolecular forces dominate, re-crystallization occurs. On the contrary, co-crystallization occurs[17]. So, in this work, we have investigated the structures and intermolecular interactions involved in the co-crystallization of HMX with DMF. The obtained results can provide important information for understanding the co-crystallization mechanism of HMX with DMF.
2 Methodology 2.1 Quantum chemistry calculationsAccording to X-ray powder diffraction experiments of β and α polymorphs of crystalline HMX (CSD code: OCHTET12 for β-HMX; OCHTET for α-HMX)[18], we have built the unit cell models. The initial geometries for single molecules are taken from the unit cells. In terms of the most possible interaction manner, the homodimers and heterodimer of α-HMX and DMF are constructed based on their optimized structures. Since the electron correlation and dispersion energy are important in these weak-interaction dimers, we apply the second-order Møller-Plesset (MP2) method and 6-31G* basis set to optimize all the molecular geometries. Stabilization energy in vacuum is calculated using supermolecular approach and corrected by counterpoise (CP) method proposed by Boys and Bernardi[19]. A self-consistent reaction field (SCRF) method with the revised Polarized Continuum Model (PCM) is used to model the system in solution. Applying the PCM model, the relative stability of α and β HMX molecules in DMF is studied. Above all calculations are performed using the Gaussian03 package[20].
2.2 Molecular dynamics simulationsThe conformation stability of HMX in DMF is explored by the MD simulation technique. The DMF solvent has the reflective index n of 1.42817, dielectric constant ε of 36.7, and density ρ of 0.9445 g·cm-3 at 298 K[21]. Different cubic boxes are constructed by the Amorphous Cell tool for different simulations. The energy minimization for the cubic box is carried out before the dynamics simulation. MD simulation is carried out in the NVT ensemble with periodic boundary conditions. The temperature control method is set to be Andersen, which chooses atom collision times from a Poisson distribution at each time step and changes their velocities according to the Boltzmann distribution.
In MD simulations, the Verlet velocity time integration method is used and initial velocities are set through the Maxwell-Boltzmann profiles at given temperatures. For potential energy calculations, the long-range Coulombic and vdW interactions are calculated by the standard Ewald method with a precision of 1×10-6[22]. All the simulations are implemented on HPZ600 workstation with Materials Studio (version 3.0)[23], using the Compass force field[24].
3 Results and Discussion 3.1 Conformation stability of HMX molecule in DMFThe HMX molecular conformation is different in its four polymorphs known as α, β, γ, and δ forms[18, 25-26]: β-HMX has a center of symmetry and the α-form possesses a crystallographic 2-fold axis, whereas the γ and δ forms obey 2-fold symmetry only approximately. The ring conformation of HMX molecule in the β-form can be described as a chair conformation[27] and will be referred to as the "β-HMX molecule" in this paper. The other conformation found in α and δ modifications of pure HMX and also believed to occur in γ-HMX is noted as the "α-HMX molecule". In Fig. 1 we give the optimized geometries of HMX and DMF. Geometry optimizations at the level of MP2/6-31G* indicate that β-HMX conformation is more stable than α-HMX by 11.14 kJ·mol-1 in vacuum. A great degree of dipolar interaction between the nitro groups can be suggested for this disparity in the stability of the two conformations. The calculated values for the dipole moment show that the α-HMX and DMF are strongly dipolar molecules, and their dipole moments are respectively 9.41 D and 4.28 D. These optimized monomers will be used to build the dimers in next section.

|
Fig.1 Optimized geometries of HMX and DMF molecules at the MP2/6-31G* level (C atom in gray, and H atom in white) |
In experiment, crystals of HMX/DMF were prepared through cooling crystallization from the warm saturated solution of β-HMX in DMF. Therefore, the theoretical model for MD simulation is constructed by placing a β-HMX molecule in the cubic simulation box containing 200 DMF molecules (2428 atoms in total). Initially, DMF molecules are placed randomly around the HMX molecule. The simulation temperature is set to be 335 K based on the experiments[1]. The time step is 1 fs with a period of 1 ns. Trajectories are collected at an interval of 500 fs for subsequent analysis. From Fig. 2, the evolving process from β-to α-form can be observed with the runtime. The conformation of the HMX molecule at 0 ps is taken from the simulation box after optimized by MM method, mainly presenting β-form characteristic. After this, in Fig. 2 (b) and Fig. 2 (c), the conformation of the HMX molecule gradually changes toward α-form due to the effects of surrounding solvent molecules. Finally, in Fig. 2 (d), the β-HMX molecule has completely converted into the α-form when the runtime reaches 650 ps.

|
Fig.2 Snapshots from the MD simulations of HMX in DMF (HMX molecule denoted as ball-stick and DMF molecules as line in gray) |
The binding energy between the HMX molecule and the surrounding solvent molecules is calculated to account for the relative stability of different conformation HMX molecules in DMF. Binding energy calculations are based on the cubic box containing 200 DMF molecules and a β-or α-HMX molecule optimized by molecular mechanics (MM) approach. MM optimization is performed using the Smart Minimizer method with "fine" convergence level. The calculated binding energy is 230.79 kJ/mol for β HMX and 301.11 kJ·mol-1 for α HMX. The relative stability of α-HMX with respect to β-HMX is calculated to be 70.32 kJ·mol-1 in DMF. Besides, quantum chemistry calculations are performed to further explore the relative stability of the HMX molecular conformation in DMF. Solvent effects are treated by the PCM model and temperature is 335 K. The total energy of a α-HMX molecule after PCM correction is -1196.640809 au at the MP2/6-31G* level, 6.81 kJ·mol-1 lower than that of the β-HMX molecule.
All the above calculations indicate that β-HMX can finally convert into α-HMX when dissolving β-HMX solid in warm DMF solvent. This explains why the HMX molecules present α-form in all the HMX/DMF crystalline forms. The geometry optimizations for β-and α-HMX and DMF molecules show that the α-HMX and DMF are strongly polar molecules, while β-HMX is nearly non-polar with dipole moment μ=0 D. The strong stability of α-HMX in DMF may be the fact that polar solvent is in favor of polar conformation molecule.
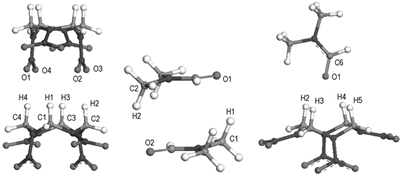
3.2 Interaction calculations between componentsAbove calculations have shown that the HMX molecule exists in the α-form in the warm DMF solution, so, we only study the interactions of α-HMX with itself and DMF in this section. In experiment, α-HMX and DMF are known to form 1: 1 solvate. In order to look into the possibility of co-crystallization of α-HMX with DMF, the homodimer and heterodimer are explored. According to the probable interaction manners, we build the structures of all possible dimers. All structures are optimized applying the MP2/6-31G* method. Obtained most stable geometries are shown in Fig. 3 and corresponding stabilization energies and hydrogen bond parameters are given in Tables 1 and 2. From the calculated stabilization energies with basis set super position error (BSSE) correction, we can find that the stabilization energy of the heterodimer α-HMX/DMF is close to that of the homodimer of α-HMX and much higher that of DMF homodimer. Besides, BSSE correction energies are much large for all dimers. According to the criterion put forward by G. R. Desiraju[28], hydrogen bonds are defined for a maximum H…O distance of 2.8 Å, a maximum distance between C and O atoms of 4.0 Å, and a minimum C—H…O angle of 110°. So, from the Table 2, we can draw that the C—H…O hydrogen bonding interactions exist in all dimers.

|
Fig.3 Dimer structures of α-HMX and DMF optimized at the MP2/6-31G* level |
| Tab.1 Energies and dipole moments for possible dimers of α-HMX and DMF in vacuum |
| Tab.2 Hydrogen bond geometries for possible dimers of α-HMX and DMF (bond length in Å, bond angle in degree) |
The hydrogen bond geometries listed in Table 2 tell us that the C—H…O interactions belong to weak H-bonds. For the DMF homodimer, due to large dispersion energy, stacked conformation is the most stable. In the α-HMX/α-HMX dimer, hydrogen donors are the methylene H atoms, and acceptors are the nitro O atoms. For the heterodimer α-HMX/DMF, the hydrogen bonds mainly occur between the DMF carbonyl O atom and four methylene H atoms of the HMX molecule. The hydrogen bond geometries are consistent with the experimental values determined in the R3c polymorph[3]. Besides, the latest experimental investigations by fourier transform infrared (FTIR), nuclear magnetic resonance (NMR), and X-ray diffraction (XRD) measures also suggested that the 1: 1 complex HMX/DMF can be formed through C—H…O hydrogen bonds[29], but did not gave the structure parameters of the C—H…O hydrogen bonds.
Above calculations show the thermodynamic stabilities of dimers and hydrogen bond geometries in vacuum. Moreover, it has verified that α-HMX can form 1: 1 complex with DMF through C—H…O hydrogen bonding. Subsequently, we apply MM method to calculate the stabilization energies of dimers in DMF and compare their relative stability. Here, we only focus on the stabilization energies of α-HMX/DMF and α-HMX/α-HMX in DMF. Placing the two dimers into the cubic simulation boxes containing 200 DMF molecules, the stabilization energies are calculated based on the optimized cubic boxes by MM method. The simulated values are 38.01 and 40.30 kJ·mol-1, respectively. We can find that the relative stability of two dimers doesn't change in solvent. Due to very close stabilization energies, the heteromeric intermolecular forces are able to compete with the homomeric intermolecular forces during the crystallization process, and co-crystallization is possible to occur. Of course, re-crystallization or co-crystallization is also related to crystalline kinetics and crystal packing arrangements. In order to further investigate this problem, a molecular dynamics for the supersaturation solution of α-HMX in DMF is conducted at 298 K and 1 atm. The reported experimental solubility of HMX is 4.35 g/100 mL DMF solvent (0.15 mol/L) at 298 K[24]. The supersaturation solution used in MD simulation contains 704 DMF and 15 HMX molecules with the concentration of 0.28 mol/L (8868 atoms in total). The solute molecules randomly distribute in solvent. The simulation box is equilibrated for 1.5 ns with periodic boundary conditions. The time step used in simulation is 1 fs.
Fig. 4 shows the final structure after equilibrated for 1.5 ns. From Fig. 4 (a), we can find that HMX molecules exist nearly as monomers without any form of aggregation, and the closest distance of HMX intermolecular atom-atom is larger than 3.9 Å, exceeding the van der Waals radii: C…N=3.07 Å, C…O=2.97 Å and O…N=2.90 Å. Therefore, the interactions between HMX molecules are probably not strong. However, C—H…O hydrogen bonding interactions can be found between α-HMX and DMF. As an example, we only illustrate interactions between a α-HMX molecule and a DMF one in Fig. 4 (b). Therefore, It is predicted that molecular self-assembly is in favor of the formation of heteromeric intermolecular forces.

|
Fig.4 Final structures after equilibrated for 1.5 ns by MD simulation[(a) HMX molecules distribution and (b) interactions between a α-HMX molecule and a DMF one in DMF supersaturation solution, this DMF molecule highlighted in stick]. The broken line denoted as the distance between the DMF carbonyl O atom and four methylene H atoms of HMX in Å, and the HMX molecule as ball-stick and DMF molecules as line |
As we known, the final crystallization form depends not only on the enthalpic properties arising from a particular kind of interaction but also on all the processes that contribute to crystal nucleation. Once nucleation of one crystal type occurs, all the solution equilibria can be displaced to favor the aggregate being trapped in the fastest growing nuclei. In general, nucleation is very difficult to model due to the requirement for both exceedingly long simulations times and large system sizes, though the successfully simulations for the nucleation of some model systems have been reported using some assumptions by MD and kinetic Monte Carlo simulations[30-31]. In this work, theoretical study for the structures and interactions involved in the crystallization process can serve an important role in explaining the co-crystallization mechanism of HMX with DMF. Clearly, further work is needed to study nucleation and crystal growth by more sophisticated computational methods such as those in development[32-35].
4 ConclusionsIn this work, we report the structures and intermolecular interactions involved in the co-crystallization of HMX with DMF. The MD simulation indicates that β-conformation will convert into α-form in DMF solvent due to stronger binding of α-HMX with surrounding solvent molecules. Quantum chemistry calculations at the level of MP2/6-31G* also suggest that α-HMX is more stable than β-HMX. This explains why all HMX molecules present α-form in the polymorphic forms of HMX/DMF solvate. The intermolecular interaction calculations indicate that heteromeric intermolecular forces is very close to the homomeric intermolecular ones, which means that co-crystallization process is possible to occur from the thermodynamics viewpoint. The intermolecular forces are mainly attributed to the C—H…O hydrogen bond interactions. Dynamics simulations for the supersaturation solution of α-HMX in DMF show that the molecular self-assembly makes for the formation of heteromeric intermolecular forces, which imply that the co-crystallization may be easier to occur compared to the re-crystallization.
| [1] |
Cobbledick R E, Small R W H. The structure of the complex formed between 1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetraazacyclooctane (HMX) and N, N-dimethylformamide (DMF)[J]. Acta Cryst, 1975. |
| [2] |
Haller T M, Rheingold A L, Brill T B. The structure of the complex between octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine (HMX) and N, N-dimethylformamide (DMF), C4H8N8O8·C3H7NO, a second polymorph[J]. Acta Cryst, 1983. |
| [3] |
Marsh R E. Converning a second polymorph of the HMX-DMF complex[J]. Acta Cryst, 1984. |
| [4] |
Jones W, Motherwell W D S, Trask A V. An emerging approach to physical property enhancement[J]. Phar Mater Sci, 2006, 31(11): 875-879. |
| [5] |
Childs S L, Chyall L J, Dunlap J T, et al. Crystal engineering approach to forming cocrystals of amine hydrochlorides with benzoic, succinic, and fumaric acids[J]. J Am Chem Soc, 2004, 126(41): 13335-13342. DOI:10.1021/ja048114o |
| [6] |
Hickey M B, Peterson M L, Scoppettuolo L A, et al. Performance comparison of a co-crystal of carbamazepine with marketed product[J]. Eur J Biopharm, 2007, 67: 112-119. DOI:10.1016/j.ejpb.2006.12.016 |
| [7] |
Trask A V, Motherwell W D S, Jones W. Pharmaceutical cocrystallization: engineering a remedy for caffeine hydration[J]. Cryst Growth Des, 2005, 5(3): 1013-1021. DOI:10.1021/cg0496540 |
| [8] |
Sun C C, Hou H. Improving mechanical properties of caffeine and methyl gallate crystals by cocrystallization[J]. Cryst Growth Des, 2008, 8(5): 1575-1579. DOI:10.1021/cg700843s |
| [9] |
Zhang J Z. Double-Base Powde[M]. Beijing: Beijing Institute of Technolgy Press, 1997, 75-76.
|
| [10] |
Selig W. Some complexes of cyclomethylenetetranitramine (HMX)[J]. Explosivstoffe, 1967, 15: 76-87. |
| [11] |
Abel J E, Marinkas P L, Bulusu S. Complex formation in RDX, HMX, TNT and some related compounds: abibliography[J]. J Ballist, 1981, 5(3): 1195-1216. |
| [12] |
Propellant made with cocrystals of cyclotetramethylenetetranitermine and ammonium perchlorate. United States Patent 40816110[P], 1978.
|
| [13] |
Shen J P, Duan X H, Luo Q P, et al. Preparation and characterization of a novel cocrystal explosive[J]. Cryst Growth Des, 2011, 11(5): 1759-1765. DOI:10.1021/cg1017032 |
| [14] |
Cruz cabeza A J, Day G M, Samuel Motherwell W D, et al. Prediction and observation of isostructurality induced by solvent incorporation in multicomponent crystals[J]. J Am Chem Soc, 2006, 128(45): 14466-14467. DOI:10.1021/ja065845a |
| [15] |
Karamertzanis P G, Price S L. Challenges of crystal structure prediction of diastereomeric salt pairs[J]. J Phys Chem B, 2005, 109(36): 17134-17150. DOI:10.1021/jp052465n |
| [16] |
Karamertzanis P G, Kazantsev A V, Issa N, et al. Can the formation of pharmaceutical cocrystals be computionally predicted? 2. Crystal structure prediction[J]. J Chem Theory Comput, 2009, 5(5): 1432-1448. DOI:10.1021/ct8004326 |
| [17] |
Aakeröy C B, Salmon D J. Building co-crystals with molecular sense and supramolecular sensibility[J]. Cryst Eng Comm, 2005, 7(72): 439-448. DOI:10.1039/b505883j |
| [18] |
Cady H H, Larson A C, Cromer D T. The crystal structure of α-HMX and a refinement of the structure of -HMX[J]. Acta Cryst., 1963, 16: 617-623. DOI:10.1107/S0365110X63001651 |
| [19] |
Boys S F, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors[J]. Mol Phys, 1970, 19(4): 553-556. DOI:10.1080/00268977000101561 |
| [20] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03, Revision D. 01, Gaussian, Inc., Wallingford CT, 2004.
|
| [21] |
Dean J A. Lange's Handbook of Chemistry, 15th ed[M]. McGrawHill, New York, 1999.
|
| [22] |
Ewald P P. The calculation of optical and electrostatic grid potential[J]. Ann Phys, 1921, 64: 253. |
| [23] |
Materials Studio 3. 0, Accelrys Inc. San Diego, CA, 2004.
|
| [24] |
Sun H. COMPASS: an ab initio force-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds[J]. J Phys Chem B, 1998, 102(38): 7338-7364. DOI:10.1021/jp980939v |
| [25] |
Choi C S, Boutin H P. A study of the crystal structure of β-cyclotetramethylene tetranitramine by neutron diffraction[J]. Acta Crys, 1970, B26: 1235-1240. |
| [26] |
Cobbledick R E, Small R W H. The crystal structure of the δ-form of 1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetraazacyclooctane (δ-HMX)[J]. Acta Cryst, 1974, B30: 1918-1922. |
| [27] |
Brill T B, Reese C O. Analysis of intra-and intermolecular interactions relating to the thermophysical behavior of alpha-, beta-, and delta-octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetraazocine[J]. J Phys Chem, 1980, 84(11): 1376. DOI:10.1021/j100448a018 |
| [28] |
Desiraju G R. Hydrogen bridge in crystal engineering: interactions without borders[J]. Acc Chem Res, 2002, 35: 565-573. DOI:10.1021/ar010054t |
| [29] |
Tian H Y, Zhang G, Wang M C, et al. The complex behavior of HMX and DMF[J]. Chinese Journal of Energetic Materials, 2009, 17(5): 541-543. |
| [30] |
Anwar J, Boateng P K. Computer simulation of crystallization from solution[J]. J Am Chem Soc, 1998, 120(37): 9600-9604. DOI:10.1021/ja972750n |
| [31] |
Piana S, Reyhani M, Gale J D. Simulating micrometre-scale crystal growth from solution[J]. Nature, 2005, 38(3): 70. |
| [32] |
Beckham G T, Peters B, Starbuck C, et al. Surface-mediated nucleation in the solid-state polymorph transformation of terephthalic acid[J]. J Am Chem Soc, 2007, 129(15): 4714. DOI:10.1021/ja0687567 |
| [33] |
Peters B, Trout B L. Obtaining reation coordinates by likehood maximization[J]. J Chem Phys, 2006, 125(5): 54108-54117. DOI:10.1063/1.2234477 |
| [34] |
Weinan E, Ren W Q, Vanden-Eijnden E. Transition pathways in complex systems: Reaction coordinates, isocommittor surfaces, and transition tubes[J]. Chem Phys Lett, 2005, 413: 242-247. DOI:10.1016/j.cplett.2005.07.084 |
| [35] |
Weinan E, Ren W Q, Vanden-Eijnden E. Finite temperature string method for the study of rare events[J]. J Phys Chem B, 2005, 109(14): 6688-6693. DOI:10.1021/jp0455430 |
The structures and intermolecular interactions for solvate HMX/DMF were ivestigated. The valuable information were given for understanding why co-crystallization occurs other than re-crystallization in DMF solution of β-HMX, and why all HMX molecules present α-form in the polymorphic forms of HMX/DMF solvate.