




高能量密度材料的开发主要是为了寻找具有更高爆炸性能、更低感度和更好热安定性的化合物[1-3]。为了满足能量和安全性的双重要求,目前国内外均希望找到高能低感的第四代含能材料——超高能量密度物质[4-6]。全氮含能化合物是近年来发展起来的一类具有良好前景的新型含能材料, 与传统的含能材料相比,全氮含能材料有着更优良的理化和爆炸性能[7-8]。因全氮含能化合物自身的优越性使其在超高能炸药、低特征信号推进剂等领域具有潜在的应用前景[9]。
国内外对全氮化合物进行了大量研究[10-12]。美国KarlChiste博士于1998年底合成出N5+, 但稳定性差,合成条件苛刻[13]。Dyke[14]等利用光谱检测到N3自由基和N3+阳离子的存在。Tobita[15]等运用密度泛函理论研究了N6异构体的振动频率、电离势等性质。Wilson[16]等对N6环与氧原子所形成的化合物进行了ab initio计算。N8、N12、N18、N20等全氮化合物目前还处于理论研究阶段[17]。理论计算表明,全氮苯N6的标准生成焓为1444.52 kJ·mol-1,爆速为14.04 km·s-1, 爆压为93.3 GPa[18],能量输出是CL-20[19-20]的3倍。本研究运用DFT-B3LYP/6-311++G(d, p)方法对氮气及全氮苯N6分子的结构进行几何优化(图 1)。运用Materials Studio程序包在7种最可几空间群(P21/c、P-1、P212121、Pbca、C2/c、P21和Pna21)中分别进行全局搜索,获得全氮苯N6的最佳分子堆积方式。分析了反应3N2(气)→N6(固)的热力学可行性,并预测其反应机理。这为全氮苯N6分子的深入研究提供了基础数据, 对指导实验合成、减少实验工作的盲目性具有重要参考价值。

|
图 1 全氮苯N6的分子结构 Fig.1 Molecular structure of the N6 |
由于B3LYP法较充分考虑电子相关,保持了从头算分子轨道(MO)方法的很多优点, 又较节省机时,采用此方法并结合6-311++G(d, p)基组,对氮气及全氮苯N6进行几何优化及频率计算,求得势能面上极小值, 振动分析无虚频,证明所得的优化构型对应于势能面上极小点。计算使用Gaussian 03量子化学软件包[21]完成。收敛精度取程序内定值。
超高能量物质多处于凝聚态尤其是晶态,固体材料比气态分子更贴近实际应用,实际存在的晶体通常具有较低的自由能(G)。根据热力学定义,
| $ G = E + pV - TS $ |
式中, E是体系的能量, J; p是压强, Pa; V是体积, m3; T是温度, K; S是熵, J·K-1。
pV项较易求出,通常其值较小, 对G的影响可忽略; 晶体在不同T时S不同,TS项对G影响较大,但因研究目的是对0 K时不同能量的堆积方式进行相对稳定性排序,故该项为0。因此能量E最低的堆积方式其G值亦最小,对应于实验中所得最稳定晶型。这是以E值大小判定可能晶型的基本依据。一般以能量最低者作为最终预测到的合理晶型。由于分子体系较大,在不同空间群中可能的堆积方式太多,故在搜索晶体结构计算E时,通常不用量子力学方法,而是运用经验性力场分子力学(MM)方法。显然,力场选择至关重要。运用Materials Studio[22]程序包中Polymorph模块和Compass[23]、Dreiding[24]两种力场,在7种最可几空间群(P21/c、P-1、P212121、Pbca、C2/c、P21和Pna21)中分别进行MM全局搜索,以求得最佳分子堆积方式。
3 结果与讨论 3.1 分子结构全氮苯N6分子是平面正六边形构型,键角均为120°,环上六个N—N键长均为0.132 nm [25],介于N=N双键(0.123 nm)与N—N单键(0.143 nm) [26]之间,表明全氮环上六个N—N键均为离域键。全氮苯N6中各个原子的电荷平均分布且基本为0, 在最高占有轨道(HOMO)和最低空轨道(LUMO)中所占的比例相同,因此各个原子的活性相同, 这与全氮苯N6高度对称的结构及等键长和键角有关。全氮苯中六个氮原子都以sp2杂化轨道互相沿对称轴的方向重叠形成六个N—N σ键,组成一个正六边形,各个氮原子中的孤对电子占据一个sp2杂化轨道,每个氮原子还有一个未杂化的p轨道,均垂直于全氮苯环平面且相互平行,每个p轨道都可以与两个相邻氮原子的p轨道侧面重叠,同时分子的骨架是闭合的,这就形成了一个闭合的共轭体系。这个体系的特点是p轨道中的π电子不再局限在两个氮原子之间,而是形成一个环状离域的π电子云,电子云平均分布在整个环上,构成两个圆形电子云,分别处于全氮苯环的上方和下方,形成了一个电子云密度完全平均化的大π键,闭合共轭体系的形成使其能量降低。
3.2 晶体结构分子模拟中的力场方法是用来计算分子结构和能量的计算方法,它通过原子核的位置来计算体系能量。对于分子动力学及分子力学方法而言,选用的力场对研究体系的适用程度是影响模拟结果的关键因素。Compass力场是“原子水平模拟研究凝聚态的分子力场”,具有较好的普适性,能够模拟小分子、高分子、金属离子、金属氧化物与金属等多种体系。它通过从头计算方法,获得分子内的键参数,同时又采用以凝聚态分子动力学为基础的经验方法优化范德华非键合参数。其准确性已通过单分子、液态分子及晶体分子共28类分子的验证[23, 27]。运用Compass和Dreiding力场在7个最可几空间群(P21/c, P-1, P212121, P21, Pbca, C2/c和Pna21)中分别进行分子力学全局搜索,以求得最佳分子堆积方式。表 1列出全氮苯N6在Compass力场下,每种空间群中具有能量极小晶型的结构参数。Dreiding力场下,全氮苯7个最可几空间群堆积方式的能量范围为13.567~13.602 kJ·mol-1·cell-1,远高于Compass力场下,各空间群堆积方式的能量。因此,Compass力场更适合全氮苯N6晶体结构的预测。由表 1可见,全氮苯N6在C2/c空间群的堆积方式具有最低能量。因此,全氮苯N6取C2/c空间群的最佳堆积方式较为合理(图 2)。
| 表 1 Compass力场下全氮苯N6搜索所得可能分子堆积方式的晶胞参数 Tab.1 Unit cell parameters of possible packing of the molecule N6 in Compass force field |

|
图 2 全氮苯N6的C2/c空间群结构 Fig.2 Molecular packing of the N6 in C2/c space group |
根据统计热力学基本原理[28]和校正后的谐振频率,求得200~800 K范围内的标准热力学函数。表 2列出氮气和全氮苯N6在不同温度下的标准恒压热容(Cp, mθ)、标准熵(Smθ)和标准焓(Hmθ)。从表 2可看出, 所有热力学函数值在200~800 K范围内均随温度的升高而增加, 这是由于在高温时振动加剧所致。对全氮苯N6不同温度下的热力学性质与温度进行关联,得到200~800 K温度范围内的Cp, mθ、Smθ、Hmθ与温度(T)函数关系分别为:
| $ \begin{array}{l} C_{{\rm{p}},{\rm{m}}}^\theta = - 0.00009\;{T^2} + 0.1783\;T + 5.3050\\ S_{\rm{m}}^\theta = - 0.00006\;{T^2} + 0.2004\;T + 155.75\\ H_{\rm{m}}^\theta = - 0.00004\;{T^2} + 0.0249\;T - 0.1470 \end{array} $ |
| 表 2 不同温度下氮气和全氮苯N6的热力学性质 Tab.2 The thermodynamic properties of nitrogen and N6 at different temperatures |
相关系数分别为0.9987、0.9999和0.9997。另外, 还可以得出dCp, mθ/dT=0.1783-0.00018T, 显然, 在200~800 K范围内温度越高,Cp, mθ随温度的变化越来越慢; 当T>990.6 K时, dCp, mθ/dT<0, Cp, mθ会随温度的升高而减小。氮气和全氮苯N6在不同温度下的Cp, mθ、Smθ和Hmθ对预测化学反应的进行提供重要的理论指导,对深入研究全氮苯N6其它热力学性质及合成可行性有一定的参考价值。
3.4 反应3N2(气)→N6(固)的可行性分析根据所得标准热力学数据,计算不同温度下反应3N2(气)→N6(固)的标准摩尔吉布斯函数变ΔrGmθ(kJ·mol-1)。根据公式ΔrGmθ=ΔrHmθ-TΔrSmθ评估了反应的热力学可行性。其中, ΔrHmθ为反应的标准摩尔生成焓; ΔrSmθ为反应的标准摩尔熵; ΔrGmθ为反应的标准摩尔吉布斯函数。
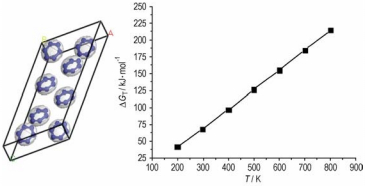
计算结果见表 3。可以看出,温度在200~800 K范围内,反应3N2(气)→N6(固)的标准摩尔生成焓随温度的升高而减小,且均小于零,反应为放热反应,升高温度,虽有利于提高反应速率,但温度升高不利于反应的进行。由于正反应为体积减小的反应,增大压强有利于反应正向进行。因此,增大压强,降低温度对反应有利。对反应3N2(气)→N6(固)不同温度下的热力学函数与温度进行关联,得到200~800 K温度范围内的标准吉布斯自由能(ΔGT)与温度(T)函数关系为如图 3所示。
| $ \Delta {G_T}=0.2895\;T - 17.839 $ |
| 表 3 反应3N2(气)→N6(固)在不同温度下的热力学函数值 Tab.3 The thermodynamic function values of reaction 3N2(g)→N6(s) at different temperatures |
相关系数为0.9998。若使反应进行,当T < 61.6 K时,ΔGT < 0。需要指出的是,我们只通过理论计算判别了反应的热力学可行性, 要进一步研究反应是否具有实用价值, 还需要考虑反应动力学即化学反应速率问题。

|
图 3 反应3N2(气)→N6(固)与温度的关系 Fig.3 Relationship for reaction 3N2(g)→N6(s) and T |
电子的重排及价键的断裂与生成是反应的主要特佂,决定了各反应状态的稳定性。氮气聚合生成全氮苯N6的反应机理分为四步(图 4)。(1)氮气分子闭壳层互相排斥。这一阶段能量的增加主要是由于分子间闭壳层的互相排斥(ΔEint)以及氮气分子自身的扭曲(ΔEdis); (2)转型的排斥能使分子发生形变,从而导致氮气分子互相靠近。该阶段扭曲能(ΔEdis)大于排斥能(ΔEint),扭曲能占主导地位,氮气分子的扭曲减小了分子闭壳层间的互相排斥; (3)键离域作用的急剧增大伴随N—N键的产生; (4) N—N键的生成,π-芳香性的形成或分子由非芳香性转化为芳香性。其中,分子几何形状及能量的变化伴随全氮苯N6分子由非芳香性向芳香性的转化。由图 5看出,过渡态Ⅱ转化为过渡态Ⅲ是反应的速决步,π键的离域作用对反应的进行起决定性作用。

|
图 4 3N2(气)→N6(固)的反应机理 Fig.4 Reaction mechanism for 3N2(g)→N6(s) |

|
图 5 反应3N2(气)→N6(固)的势能曲线 Fig.5 Potential energy curve of reaction 3N2(g)→N6(s) |
由于N≡N结合牢固,若在N≡N上引入高能吸电子基团(如—NO2,—CN等),减弱氮氮间的电子云密度,降低氮氮间的稳定性,从而降低反应的势垒。吸电子能力越强,反应势能越小。同时,反应中心(叁键)电子云密度越低,闭壳层的排斥能(ΔEint)越小,使扭曲能成为主导,增加了p键的离域作用,有利于反应的进行。
4 结论采用DFT-B3LYP/6-311++G(d, p)方法,计算获得了氮气和全氮苯N6分子的最优化构型及热力学性质,判断了反应3N2(气)→N6(固)的热力学可行性,并对反应机理进行了阐述。结果表明,全氮苯N6分子形成了一个闭合的共轭体系,大大降低了体系的能量。全氮苯N6的最佳堆积方式为C2/c空间群,晶体密度为1.949 g·cm-3。反应温度T < 61.6 K时,3N2(气)→N6(固)是可行的。π键离域作用的急剧增大对过渡态活化能的高低有直接影响,其中,扭曲能的增加是π键离域作用增大的关键。由于氮气的高稳定性,寻求一种有效的催化剂以降低其反应的活化能,有助于反应3N2(气)→N6(固)的进行。
| [1] |
Wei T, Zhu W H, Zhang J J, et al. DFT study on energetic tetrazolo-[1, 5-b]-1, 2, 4, 5-tetrazine and 1, 2, 4-triazolo-[4, 3-b]-1, 2, 4, 5-tetrazine derivatives[J].
Journal of Hazardous materials, 2010, 179(1): 581-590. |
| [2] |
Joo Y H, Twamley B, Shreeve J M. Carbonyl and oxalyl bridged bis(1, 5-diamino tetrazole)-based energetic salts[J].
Chemistry-A European Journal, 2009, 15(36): 9097-9104. DOI:10.1002/chem.v15:36 |
| [3] |
李洪珍, 周小清, 王述存, 等. HNIW在乙酸乙酯-正庚烷溶剂体系中的结晶机制研究[J].
含能材料, 2012, 20(1): 30-34. LI Hong-zhen, ZHOU Xiao-qing, WANG Shu-cun, et al. Crystallization mechanism of ε-HNIW in ethyl acetate/n-heptane solution[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2012, 20(1): 30-34. |
| [4] |
Song X L, Li J C, Hou H, et al. Extensive theoretical studies of a new energetic material: tetrazino-tetrazine-tetraoxide (TTTO)[J].
Journal of Computational Chemistry, 2009, 30(12): 1816-1820. DOI:10.1002/jcc.v30:12 |
| [5] |
Cui J, Zhang Y, Zhao F J, et al. HB(N-5)(3)M (M = Li, Na, K, and Rb): A new kind of pentazolides as HEDMs[J].
Progress in Natural Science, 2009, 19(1): 41-45. DOI:10.1016/j.pnsc.2008.06.010 |
| [6] |
Zhou H, Wong N B, Lau K C, et al. Discovery of singlet diradicals: theoretical study on the cage species C14N12H6 and its six derivatives[J].
Journal of Physical Chemistry A, 2007, 111(39): 9838-9847. DOI:10.1021/jp072460l |
| [7] |
Zarko V E. Searching for ways to create energetic materials based on polynitrogen compounds[J].
Combustion Explosion and Shock Waves, 2010, 46(2): 121-131. DOI:10.1007/s10573-010-0020-x |
| [8] |
Noyman M, Zilberg S, Haas Y. Stability of polynitrogen compounds: the importance of separating the sigma and pi electron systems[J].
Journal of Physical Chemistry A, 2009, 113(26): 7376-7382. DOI:10.1021/jp811508j |
| [9] |
李辉, 王伯周, 于倩倩, 等. 3, 3'-二(四唑-5-基)二呋咱基醚的合成与表征[J].
含能材料, 2012, 20(1): 18-21. LI Hui, WANG Bo-zhou, YU Qian-qian, et al. Synthesis and characterization of 3, 3'-bis(tetrazol-5-yl)difurazanyl ether[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2012, 20(1): 18-21. |
| [10] |
Wang L J, Warburton P, Mezey P G. Theoretical prediction on the synthesis reaction pathway of N6 (C2h)[J].
Journal of Physical Chemistry A, 2002, 106(11): 2748-2752. DOI:10.1021/jp0141833 |
| [11] |
Tobita M, Bartlett R J. Structure and stability of N6 isomers and their spectroscopic characteristics[J].
Journal of Physical Chemistry A, 2001, 105(16): 4107-4113. DOI:10.1021/jp003971+ |
| [12] |
Gagliardi L, Evangelisti S, Barone V, et al. On the dissociation of N6 into 3N2 molecules[J].
Chemical Physics Letters, 2000, 320(5): 518-522. |
| [13] |
Christe K, Wilson W, Sheehy J, et al. N5+: A novel homoleptic polynitrogen ion as a high energy densitymaterial[J].
Angewandte Chemie International Edition, 1999, 38(13-14): 2004-2009. DOI:10.1002/(ISSN)1521-3773 |
| [14] |
Dyke J M, Jonathan N B H, Lewis A E, et al. Vacuum ultraviolet photoelectron spectroscopy of transient species. Part 15. The N3(X2Ⅱ) radical[J].
Molecular Physics, 1982, 47(5): 1231-1240. DOI:10.1080/00268978200100922 |
| [15] |
Tobita M, Bartlett R J. Structure and stability of N6 isomers and their spectroscopic characteristics[J].
Journal of Physical Chemistry A, 2001, 105(16): 4107-4113. DOI:10.1021/jp003971+ |
| [16] |
Wilson K J, Perera S A, Bartlett R J. Stabilization of the pseudo-benzene N6 ring with oxygen[J].
Journal of Physical Chemistry A, 2001, 105(32): 7693-7699. DOI:10.1021/jp010783q |
| [17] |
Dixon D A, Feller D, Christe K O, et al. Enthalpies of formation of gas-phase N3, N3-, N5+, and N5-from ab initio molecular orbital theory, stability predictions for N5(+)N3(-) and N5(+)N5(-), and experimental evidence for the instability of N5(+)N3(-)[J].
Journal of the American Chemical Society, 2004, 126(3): 834-843. DOI:10.1021/ja0303182 |
| [18] |
Talawar M B, Sivabalan R, Asthana S N, et al. Novel ultrahigh-energy materials[J].
Combustion, Explosion, and Shock Waves, 2005, 41(3): 264-277. DOI:10.1007/s10573-005-0031-1 |
| [19] |
宋振伟, 严启龙, 李笑江, 等. 溶剂中ε-CL-20的晶型变化[J].
含能材料, 2010, 18(6): 648-653. SONG Zhen-wei, YAN Qi-long, LI Xiao-jiang, et al. Crystal transition of ε-CL-20 in different solvent[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2010, 18(6): 648-653. |
| [20] |
Makarov A, LoBrutto R, Christodoulatos C, et al. The use of ultra high-performance liquid chromatography for studying hydrolysis kinetics of CL-20 and related energetic compounds[J].
Journal of Hazardous Materials, 2009, 162(2-3): 1034-1040. DOI:10.1016/j.jhazmat.2008.05.157 |
| [21] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03[CP], Gaussian, Inc. : Pittsburgh PA, 2003.
|
| [22] |
Materials Studio Blends Module, Version 4. 4; software for miscibility estimation: Theory in Blends[CP], Accelrys Software Inc. : San Diego, CA, 2008.
|
| [23] |
Sun H. Compass: An ab initio force-field optimized for condense-phase applications-over-view with details on alkanes and benzene compounds[J].
Journal of Physical Chemistry B, 1998, 102(38): 7338-7364. DOI:10.1021/jp980939v |
| [24] |
Mayo S L, Olafson B D, Goddard W A. Dreiding: A generic forcefield for molecular simulations[J].
Journal of Physical Chemistry, 1990, 94(26): 8897-8909. DOI:10.1021/j100389a010 |
| [25] |
吴文胜, 吴思展, 舒华, 等. 六氮苯芳香性的量子化学计算研究[J].
广州化工, 2006, 34(4): 23-26. WU Wei-sheng, WU Si-zhan, SHU Ha, et al. Research on DFT of the aromaticity on the pseudo-benzene N6 ring[J]. Guangzhou Chemical Industry, 2006, 34(4): 23-26. |
| [26] |
Lin M H.
Calculation model and application of quantum chemistry[M]. Beijing: Science and Technology Press, 2004 |
| [27] |
Sun H. Ab initio calculations and force field development for computer simulation of polysilanes[J].
Macromolecules, 1995, 28(3): 701-712. DOI:10.1021/ma00107a006 |
| [28] |
Hill T L.
Introduction to statistical thermodynamics[M]. New York: Addision-Wesley Publishing Company, 1960 |
Fully optimized calculation and vibrational analysis for nitrogen and pseudo-benzene N6 were carried out using density-functional theory (DFT) method at B3LYP/6-311++G(d, p) level. Molecular mechanics (MM) method with Compass and Dreiding force fields was used to predict molecular packing for N6 among the 7 most probable space groups (P21/c, P-1, P212121, P21, Pbca, C2/c, and Pna21), respectively. The feasibility of process was analyzed with thermodynamics and reaction mechanism was predicted.