呋咱环是构建含C、H、O、N高能量密度化合物的一种有效的结构单元。呋咱类化合物分子结构中含有大量的C—N、C=N键和N=N键,使其具有如下优点:(1)标准生成焓高;(2)氮氧含量高;(3)能量密度高;(4)具有芳香性,分子热力学稳定性较好;(5)熔点较低等[1-5]。以上优良的性能使得呋咱化合物成为国内外含能材料研究的热点,而将呋咱环与芳环或其它高氮环如三唑环[6-9]、四唑环进行有效的的结合是构建新型含能材料的新思路,此举必将大大拓展呋咱类含能化合物的研究和应用空间:将多硝基芳环引入到呋咱化合物中,分子量增大且呋咱环上的氨基与邻近的硝基形成氢键,提高了这类化合物的熔点和热稳定性,如3-硝基-4-苦基氨基呋咱熔点达到181 ℃;三唑环、四唑环具有下列特点:(1)含有大量的N=N、C—N键及大的环张力,具有非常高的正生成焓[10-11];(2)低的碳氢含量易于实现氧平衡;(3)分解产物大都为清洁的氮气,是绿色环境友好型含能材料;(4)具有芳香性,稳定性好。将它们与呋咱环相连形成的化合物更能凸显以上性质,有望作为高能钝感炸药。
Coburn M D[12]首次报道了3-(N-2, 4, 6-三硝基苯基)-氨基-4-硝基呋咱的合成:3, 4-二氨基呋咱先与2, 4, 6-三硝基氯苯反应生成3-(N-2, 4, 6-三硝基苯基)-氨基-4-氨基呋咱,然后用90%的双氧水氧化氨基得到3-(N-2, 4, 6-三硝基苯基)-氨基-4-硝基呋咱,总合成产率为13%;Finnegan WG[13]曾经报道了氰基与叠氮化钠、氯化铵成四唑环的这类反应,指出对于生成5位取代的四唑环,在一系列铵盐中以叠氮化铵产率最高,同时也指出较高沸点的DMF是这类反应最适合的溶剂;王伯周[14]等人于2011年首次报道3-硝基-4-(1H-5-四唑基)呋咱的合成,用50%的双氧水、浓硫酸及钨酸钠体系做氧化剂将3-氨基-4-(1H-5-四唑基)呋咱氧化成3-硝基-4-(1H-5-四唑基)呋咱;Leonard P W[15]等人于2010年报道了3, 3′-(1H-5-四唑基)-4, 4′-偶氮呋咱的合成;Vladimir Yu[8]等人于2004年提出了3-氨基-4-(4-氰基-5-氨基-1-1,2,3-三唑基)呋咱的合成路线,但是没指出具体的合成过程。以上文献报道的方法存在产率不理想和反应条件苛刻的弊端,反应试剂(如90% H2O2)不易制取或后处理(如除去DMF)相对困难,针对以上问题本研究提出了合成这五种化合物的新方法,方便快捷、改善了反应条件、提高了产率。如本研究在合成N-(2,4,6-三硝基苯基)-3-氨基-4-硝基呋咱的过程中避免了使用浓度较高的双氧水;用30%的双氧水代替90%的双氧水合成3-硝基-4-(1H-5-四唑基)呋咱;用无水乙醇代替沸点高的DMF制得3-氨基-4-(1H-5-四唑基)呋咱等。
2 实验部分 2.1 实验试剂与仪器3-氨基-4-硝基呋咱(自制,>98%)、3-氨基-4-氰基呋咱(自制,>98%)、3-氨基-4-叠氮基呋咱(自制,>98%)、2, 4, 6, -三硝基氯苯(自制,>98%)、氯化铵(AR, 成都市科龙化工试剂厂)、三乙胺(AR, 国药集团化学试剂有限公司)、叠氮化钠(工业纯,浙江金华器材有限公司)、浓盐酸(AR, 上海凌峰化学试剂有限公司)、高锰酸钾(AR, 上海凌峰化学试剂有限公司)、30%双氧水(AR, 南京化学试剂有限公司)、钨酸钠(AR, 上海华精生物高科技有限公司)、甲磺酸(AR, 成都市科龙化工试剂厂)、碳酸钾(AR, 成都市科龙化工试剂厂)、丙二腈(CP, 国药集团化学试剂有限公司)。
Bruker Avance Ⅲ 500M数字化核磁共振仪(瑞士Bruker公司)、TSQ Quantum质谱仪(美国Finnigan公司)、Bruker Tensor 27傅里叶红外变换光谱仪(瑞士Bruker公司)、LC-20AT高效液相色谱(日本岛津)。
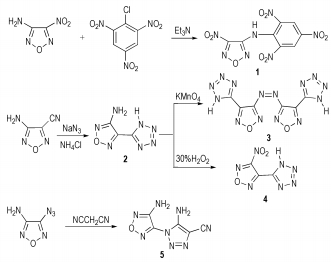
2.2 合成路线几种呋咱衍生物的合成路线如下图 1所示。

|
图 1 目标化合物的合成路线 Fig.1 Synthesis routes of target compounds |
称取3-氨基-4-硝基呋咱0.33 g(2.50 mmol),加入无水N, N-二甲基甲酰胺4 mL,得黄色溶液。向反应体系中加入7滴三乙胺,溶液逐渐变为暗红色。滴加完毕后加入0.63 g(2.55 mmol) 2, 4, 6-三硝基氯苯(苦基氯),慢慢升温至55 ℃,反应约半小时。反应体系中依次加入25 mL乙醇,50 mL蒸馏水,反应液渐渐变成黄色并产生黄色粉末状沉淀,静置、过滤、真空干燥得黄色固体0.26 g,产率36.1%。13C NMR(DMSO-d6): δ 123.30, 128.95, 140.49, 144.18(苯环上四个碳的化学位移); 152.98, 155.27(呋咱环上两个碳的化学位移)。ESI-MS, 一级质谱, m/z: 339.80[M-H]-; 二级质谱, m/z: 339.80[M-H]-, 247.85[M-H-NO2-NO2]-, 201.89[M-H-NO2-NO2-NO2]-。
2.3.2 3-氨基-4-(1H-5-四唑基)呋咱23-氨基-4-氰基呋咱0.11 g(1.00 mmol)、叠氮化钠0.078 g(1.20 mmol)、氯化铵0.064 g(1.20 mmol)及5 mL无水乙醇混合于三口烧瓶中,磁力搅拌器搅拌,加热回流,20 min后停止反应。过滤,滤液除去溶剂乙醇,得白色固体;滤渣用乙醇洗脱数次,过滤,滤液除去乙醇,合并两部分固体得3-氨基-4-(1H-四唑基)呋咱0.14 g,产率90.9%。1H NMR(DMSO-d6): δ 6.18(s, 2H, NH2), 9.98(s, H, NH)。13C NMR(DMSO-d6): δ 135.68, 146.92, 154.81。IR(KBr, υ/cm-1): 3425, 3324, 3255, 1690, 1630, 1393, 1150, 1034, 981, 781。ESI-MS, 一级质谱, m/z: 152.05[M-H]-; 二级质谱, m/z: 151.54[M-H]-, 119.94[M-H-N3]-, 93.98[M-H-N3-O]-, 92.87[M-H-N3-O-H]-。
2.3.3 3, 3′-(1H-5-四唑基)-4, 4′-偶氮呋咱3常温下,7.5 mL乙腈中加入3-氨基-4-(1H-5-四唑基)呋咱0.50 g(3.27 mmol),再向反应容器中加入7.5 mL 35%的盐酸,固体完全溶解得微黄色溶液。缓慢加入KMnO4 0.5 g / H2O 7.5 mL,加一滴时反应液变成淡绿色,继续滴加变成深绿色,最后变成紫色。滴加完毕搅拌30 min,反应液最终变成棕色。滴加数滴5% H2O2,反应体系变成黄色并有黄色沉淀产生,于0 ℃左右静置,过滤。滤饼用饱和碳酸钾溶夜处理,过滤,滤液用盐酸酸化,冷却重新析出沉淀。过滤,干燥得黄色粉末固体0.28 g,产率86.8%。1H NMR(DMSO-d6): δ 13.83(s, H, NH)。13C NMR(DMSO-d6): δ 146.22, 140.06, 161.26。IR(KBr, υ/cm-1): 3431, 1634, 1529, 1474, 1320, 1163, 1059, 1000, 767。ESI-MS, 一级质谱, m/z: 300.89[M-H]-; 二级质谱, m/z: 300.87[M-H]-, 257.73[M-H-N3-H]-, 189.99[M-H-N3-H-N4H]-, 161.86[M-H-N3-H-N4H-N2]-。
2.3.4 3-硝基-4-(1H-5-四唑基)呋咱41.65 g Na2WO4加入到12 mL 30%的双氧水中,得绿色溶液。0.55 g(3.6 mmol) 3-氨基-4-(1H-5-四唑基)呋咱溶于3 mL CH3SO3H后加入到上述绿色溶液中,维持滴加温度0~5 ℃。溶液变成无色,常温下反应1 h变成海洋蓝色,继续反应最后变成浅黄绿色悬浮液。反应完毕后过滤,滤液依次经乙酸乙酯萃取,水洗,硫酸镁干燥,减压蒸馏除去溶剂得类白色固体0.51 g,产率75.6%。1H NMR(DMSO-d6): δ 11.66(s, H, NH)。13C NMR(DMSO-d6): δ 158.95, 146.38, 140.22。IR(KBr, υ/cm-1): 3090, 1622, 1563, 1472, 1397, 1328, 1305, 1146, 1115, 996, 826。ESI-MS, 一级质谱, m/z: 181.96[M-H]-; 二级质谱, m/z: 181.91[M-H]-, 154[M-H-CN4]-。IR(KBr, υ/cm-1): 3090, 1622, 1563, 1472, 1397, 1328, 1305, 1146, 1115, 996, 826。
2.3.5 3-氨基-4-(4-氰基-5-氨基-1-1,2,3-三唑基)呋咱5于50 mL圆底烧瓶中加入3-氨基-4-叠氮基呋咱0.2 g(1.59 mmol),丙二腈0.14 g(2.12 mmol),20 mL水。加热到40~45 ℃,得透明溶液,加入少量碳酸钾,反应体系立即产生大量固体。升温到55 ℃反应20 min,静置、过滤、真空干燥得粉紫色固体0.28 g,产率91.8%。1H NMR(DMSO-d6): δ 6.63(s, 2H, NH2), 7.80(s, 2H, NH2)。IR(KBr, υ/cm-1): 3460, 3330, 3256, 3187, 2246, 1635, 1582, 1480, 1382, 1257, 1009, 729。ESI-MS, 一级质谱, m/z: 190.99[M-H]-; 二级质谱, m/z: 190.95[M-H]-, 132.05[M-H-CHN4]-。
3 结果与讨论 3.1 3-(N-2, 4, 6-三硝基苯基)-氨基-4-硝基呋咱3-氨基-4-硝基呋咱与2, 4, 6-三硝基氯苯的取代反应,副产物较多,产率相对较低。可能的原因有以下几点:(1) 2, 4, 6-三硝基氯苯中存在的三个硝基的强吸电子作用使得反应体系中的其他的亲核试剂进攻与氯相连的碳原子,特别是OH-进攻生成2, 4, 6-三硝基苯酚非常容易发生。(2)反应的空间位阻较大,三个硝基基团的存在阻碍亲核试剂3-氨基-4-硝基呋咱的进攻。
首先将3, 4-二氨基呋咱转变成3-氨基-4-硝基呋咱,再与2, 4, 6-三硝基氯苯发生取代反应(先氨基氧化为硝基,再取代),相较于引言中Michael D.Coburn[12]的方法(先取代,再氨基氧化为硝基):避免使用不易制取且危险的90%双氧水,同时避免了因先取代而出现的空间位阻问题,安全,方便,产率(总的合成产率24%)相对较高(文献总产率13%)。
3.2 3-氨基-4-(1H-5-四唑基)呋咱该化合物中氰基的反应活性较高,反应可在较低温度进行,不需要采用沸点高的DMF[13]做溶剂,选择沸点相对较低的乙醇回流即可,后处理相对简单。叠氮化钠和氯化铵稍微过量以保证原料全部转化。
3.3 3, 3′-(1H-5-四唑基)-4, 4′-偶氮呋咱以单电子氧化剂高锰酸钾将3-氨基-4-(1H-5-四唑基)呋咱中的氨基转变成3, 3′-(1H-5-四唑基)-4, 4′-偶氮呋咱,选择性较高,没有副反应发生,产率高。反应最后加入双氧水是为了除去过量的高锰酸钾,便于观察产物的颜色。
3.4 3-硝基-4-(1H-5-四唑基)呋咱采取30%双氧水、甲烷磺酸及钨酸钠体系做氧化剂:30%的双氧水较50%的双氧水[15]更安全、更廉价、更易制取;甲烷磺酸的酸性比硫酸的酸性较弱,更易控制反应的温度和速度。若仅用甲磺酸与双氧水的氧化体系氧化未得到目标化合物,加入钨酸钠后则可以得到,因为双氧水直接与有机物反应的活性较低,酸性条件下加入钨酸钠活化双氧水:一方面钨酸钠与双氧水形成过钨酸配合物,在催化反应中作为助催化剂;另一方面钨酸钠成弱碱性,加入甲磺酸反应体系的pH相对降低,氧化体系呈酸性,提高双氧水的氧化能力[16]。
3.5 3-氨基-4-(4-氰基-5-氨基-1-1,2,3-三唑基)呋咱3-氨基-4-叠氮基呋咱与丙二腈反应为一个1, 3-偶极环加成反应:由于3-氨基-4-叠氮基呋咱在冷水中溶解度较差,故将其加入水中升温到40 ℃左右使原料全部溶解,再进行后续反应,否则产率较低;该反应不加碳酸钾时进行很缓慢,加入碳酸钾为反应提供碱性环境,起催化作用,加入时反应现象相当明显,立即产生大量紫红色的固体。
4 结论对五种呋咱化合物的合成方法在反应条件如温度、试剂或溶剂等方面进行了改进,反应条件相对温和,产率相对提高:以3-氨基-4-硝基呋咱与苦基氯于55 ℃左右在溶剂DMF中发生取代反应合成了3-(N-2, 4, 6-三硝基苯基)-氨基-4-硝基呋咱;叠氮基与3-氨基-4-氰基呋咱在乙醇回流条件下发生[3+2]环加成反应合成90.9%的3-氨基-4-(1H-5-四唑基)呋咱,并在此基础上一方面以高锰酸钾为氧化剂常温下进行偶联合成出3, 3′-(1H-5-四唑基)-4, 4′-偶氮呋咱,另一方面用30%双氧水、钨酸钠及甲磺酸体系为氧化剂于低温下氧化氨基制得3-硝基-4-(1H-5-四唑基)呋咱,两者的产率分别为86.8%和75.6%;3-氨基-4-叠氮基呋咱与丙二腈在碳酸钾的催化下于55 ℃左右发生1.3-偶极环加成反应以91.8%的产率制得3-氨基-4-(4-氰基-5-氨基-1-1,2,3-三唑基)呋咱。
| [1] |
WANG Jun, LI Jin-shan, DONG Hai-shan, et al. A novel insensitive high explosive 3, 4-bis(aminofurazano) furoxan[J].
Propellants, Explosives, Pyrotechnics, 2008, 33(5): 347-352. DOI:10.1002/prep.v33:5 |
| [2] |
Kakanejadifard A, Farnia S M, Golamreza N. A modified-one pot synthesis of diaminoglyoxime[J].
Iran J Chem & Chem Eng, 2004, 23(1): 117-118. |
| [3] |
李战雄, 唐青松, 欧育湘, 等. 呋咱含能衍生物合成研究进展[J].
含能材料, 2002, 10(2): 59-65. LI Zhan-xiong, TANG Qing-song, OU Yu-xiang, et al. Synthesis status of furazanno energetic derivatives[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2002, 10(2): 59-65. |
| [4] |
范艳洁, 王伯周, 周彦水, 等. 3, 3'-二氰基-4, 4'-偶氮呋咱(DCAF)合成及晶体结构[J].
含能材料, 2009, 17(4): 385-388. FAN Yan-jie, WANG Bo-zhou, ZHOU Yan-shui, et al. Synthesis and crystal strecture of 3, 3'-dicyano-4, 4'-azofurazan(DCAF)[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(4): 385-388. |
| [5] |
李洪珍, 周小清, 李金山, 等. 3-氨基-4-硝基呋咱和3, 3'-二硝基-4, 4'-偶氮呋咱的合成研究[J].
有机化学, 2008, 28(9): 1646-1648. LI Hong-zhen, ZHOU Xiao-qing, LI Jin-shan, et al. Synthesis of 3-amino-4-nitrofurazan and 3, 3'-dinitro-4, 4'-azofurazan[J]. Chinese Journal of Organic Chemistry, 2008, 28(3): 1646-1648. |
| [6] |
Rozhkov V Y, Batog L V, Struchkova M I. Nucleophilic substitution in the series of(1, 2, 3-triazol-1-yl)-1, 2, 5-oxadiazoles. Reactions with N-, O-, and S-nucleophiles[J].
Russian Chemical Bulletin, 2005, 54(8): 1923-1934. DOI:10.1007/s11172-006-0059-8 |
| [7] |
Batog L V, Rozhkov V Y, Struchkova M I. Azido-1, 2, 5-oxadiazoles in reactions with 1, 3-dicarbonyl compounds[J].
Mendeleev Commun, 2002, 12(4): 159-161. DOI:10.1070/MC2002v012n04ABEH001589 |
| [8] |
Rozhkov V Y, Batog L V, Shevtsova E K, et al. Synthesis and dimroth rearrangement of 3-amino-4-(5-amino-1H-1, 2, 3-triazol-1-yl)-1, 2, 5-oxadiazoles[J].
Mendeleev Commun, 2004, 14(2): 76-77. DOI:10.1070/MC2004v014n02ABEH001891 |
| [9] |
Batog L V, Rozhkov V Y, Strelenko Y A, et al. Synthesis of(1, 2, 3-triazol-1-yl) furazans. 2. Reaction of azidofurazans with morpholinonitroethene[J].
Chemistry of Heterocyclic Compounds, 2000, 36(3): 343-345. DOI:10.1007/BF02256874 |
| [10] |
刘晓建, 张慧娟, 庞思平, 等. 唑类含能离子化合物的合成研究进展[J].
火炸药学报, 2010, 33(1): 6-10. LIU Xiao-jian, ZHANG Hui-juan, PANG Si-ping, et al. Progress of study on the synthesis of azole energetic ionic compounds[J]. Chinese Journal of Explosives & Propellants, 2010, 33(1): 6-10. |
| [11] |
Klapötke TM, Sproll SM. Nitrogen-rich polymers based on 5-bromo-1-vinyl-1H-tetrazole[J].
European Journal of Organic Chemistry, 2010(6): 1169-1175. |
| [12] |
Coburn MD. Picrylamino-substituted heterocycles. Ⅱ. Furazans[J].
Journal of Heterocyclic Chemistry, 1968, 5(1): 83-87. DOI:10.1002/jhet.v5:1 |
| [13] |
Finnegan WG, Henry RA, Lofquist R. An improved synthesis of 5-substituted tetrazoles[J].
Journal of the American Chemical Society, 1958, 80(15): 3908-3911. DOI:10.1021/ja01548a028 |
| [14] |
WANG Bo-zhou, ZHANG Guo-fang, HUO Huan, et al. Synthesis, characterization and thermal properties of energetic compounds derived from 3-amino-4-(tetrazol-5-yl)furazan[J].
Chinese Journal of Chemistry, 2011, 29(5): 919-924. DOI:10.1002/cjoc.201190189 |
| [15] |
Leonard PW, Chavez DE, Pagoria PF, et al. Azotetrazolylfurazan and nitrogenous salt derivatives[J].
Propellants, Explosives, Pyrotechnics, 2010, 36(3): 233-239. |
| [16] |
张科良, 乔小安, 屈撑囤, 等. 季膦盐和钨酸钠催化下双氧水溶液清洗清洁氧化醇和烯烃研究[J].
西安石油大学学报(自然科学版), 2008, 23(1): 77-80. ZHANG Ke-liang, QIAO Xiao-an, QU Cheng-dun, et al. Oxidation of alcohol and olefins using hydrogen peroxide solution as oxidant under the catalysis of quaternary phosphonium and sodium tungstate[J]. Journal of Xi'an Shiyou University(Natural Science Edition), 2008, 23(1): 77-80. |