




2. 中国工程物理研究院化工材料研究所, 四川 绵阳 621900
2. Institute of Chemical Materials, CAEP, Mianyang 621900, China
近年来, 越来越多研究者开始关注环境媒介效应对化学过程的影响, 其中最重要的一个方面是溶液环境中的溶剂化效应。对于一种或几种溶液中的溶质而言, 由于溶剂化效应的影响, 溶质分子的几何结构、电子结构、能量以及分解过程等都会表现出与气态分子不尽相同的状态, 很多科研工作者对于以上问题都进行了较为系统的研究, 并得到了很多有意义的结论[1-12]。
无论是单质炸药还是高聚物粘结炸药其制备过程要接触到多种溶剂。如柴涛等[13]利用恒容静态平衡法, 测定了包覆用氟橡胶-乙酸乙酯-
本研究针对体系为含CHON轻原子体系, 密度泛函方法如Becke型3参数密度泛函模型, 此模型采用Lee-Yang-Parr泛函, B3LYP方法常能给出合理几何结构参数和电子结构参数, 如姬广富[19]在B3LYP/6-31水平上对TATB进行几何优化, 并得到和实验值十分一致的键长、键角数据。由于体系含氢, 因此在基组选择上考虑了轻原子的弥散和极化, 本文在B3LYP/6-311++G(d, p)[20, 21]水平上对全部22种炸药分子进行了气态几何优化, 22种炸药分别为: 2, 4, 6, 8, 10, 12-六硝基-2, 4, 6, 8, 10, 12-六氮杂伍兹烷(CL-20)、2, 4, 6-三硝基甲苯(TNT)、三硝基苯(TNB)、硝化甘油(NG)、硝基甲烷(NM)、1, 3, 5, 7-四硝基金刚烷(TNA)、3-硝基-1, 2, 4-三吡咯-5-酮(NTO)、2, 4-二硝基咪唑(2, 4DNI)、环三甲撑三硝胺(RDX)、2, 6-二氨基-3, 5-二硝基吡嗪-1-氧化物(LLM105)、二硝基甘脲(DINGU)、1, 1-二氨基-2, 2-二硝基乙烯(FOX7)、1, 3, 3-三硝基氮杂环丁烷(TNAZ)、硝基胍(NQ)、环四甲撑四硝胺(HMX)、四硝基立方烷(TNC)、硝基乙烷(NE)、2, 4, 6-三硝基苯酚(PA)、二乙醇硝胺二硝酸酯(DINA)、4, 10-二硝基-2, 6, 8, 12-四氧杂-4, 10-二氮杂四环[5.5.0.05, 903, 11]十二烷(TEX)。图 1为各炸药分子优化后的气态平衡结构。

|
图 1 各炸药分子气态平衡结构 Fig.1 Gaseous equilibrium structures of the explosive molecules |
为考察溶剂化效应对各炸药分子的影响, 对优化的结果进行同水平PCM[22]计算, 选取7种有代表性的溶剂:苯(BEN)、甲苯(TOL)、二氯甲烷(DCM)、二氯乙烷(DCE)、丙酮(ACE)、二甲基亚砜(DMSO)和水(W), 用UFF力场考虑7种溶剂半径, 7种溶剂的偶极矩计算结果以及介电常数列于表 1。
| 表 1 7种溶剂的介电常数和偶极矩 Tab.1 Dielectric constants(EPS) and dipole moments of the seven solvents |
为考察各炸药特征键的键离解能受溶剂化效应的影响, 对硝基以及全部22种炸药分子离去硝基后的结构进行了同水平的开壳层计算。为确定驻点为局域最小以及进行零点能校正, 对所有计算的结构都进行了频率分析。所有计算用GAUSSIAN 03[23]软件包完成。
3 结果与讨论 3.1 溶剂化效应对炸药分子结构的影响 3.1.1 溶剂化效应对炸药分子特征键键长的影响所谓炸药分子的特征键, 指的是炸药分子中最弱的键, 通常是与硝基连接的共价键, 特征键的键能会对炸药的分解产生较为直接的影响, 而键长是衡量键能的一项重要指标。各炸药分子在不同溶剂中的特征键长列于表 2, 可见, 在溶液中, 几乎所有的炸药的特征键的键长都较气态有所减小。
| 表 2 炸药分子在7种溶剂中的特征键键长 Tab.2 Feature bond lengths of the explosive molecules in seven solvents |
任意一种炸药在介电常数相近的苯和甲苯中的特征键长很相近, 甲苯中的键长要略小于苯, 变化不超过0.8%, 在介电常数相近的二氯甲烷以及二氯乙烷中也存在类似结果。而在介电常数相差较大的苯与二氯甲烷中特征键键长的差别比较明显, 这说明特征键键长的变化要受到溶剂介电常数的影响, 也就是说, 特征键键长的缩短是受溶剂极性影响的。而在介电常数相差很大的丙酮、二甲基亚砜以及水中, 这些炸药分子的特征键长并没有太大变化, 说明特征键键长受溶剂极性的影响是有上限的。图 2为7种溶剂中炸药分子的特征键长变化趋势图, 可见每种炸药的特征键长互有差别, 但是它们在不同溶剂中的特征键变化趋势大体一致, 这意味着可用其中一种炸药分子的特征键长在不同溶剂中的变化趋势来定性表征这里所讨论的全部炸药分子。

|
图 2 7种溶剂中炸药分子的特征键长变化趋势 Fig.2 Change trend of the feature bonds lengths of the explosive molecules in seven solvents |
下面仅以NQ为例, 给出它在7种溶剂中键长的变化曲线, 如图 3所示。其中横坐标0点对应的值为气态分子的特征键键长。可见在介电常数为小于丙酮(10.36)的溶剂, 其键长随介电常数变化较大, 特征键键长受极性的影响较大, 介电常数大于丙酮(10.36)的溶剂,其特征键键长变化趋于平缓。

|
图 3 NQ特征键长在7种溶剂中的变化曲线 Fig.3 Change curve of the feature bond length of NQ in seven solvents |
硝基电荷, 即最弱键所连接的硝基上两个氧原子以及一个氮原子的电荷加和, 已经被证明和感度有很好的相关度, 是评价炸药安全性的一个重要指标[24]。所有计算的炸药分子在7种溶剂中的硝基电荷值列于表 3。图 4所示为7种溶剂中炸药分子的硝基电荷变化趋势。可见, 加入溶剂后, 绝大多数炸药的硝基电荷都会明显增大, 而随着溶剂介电常数的变大, 硝基电荷增大的量也会随之增大, 这说明溶剂极性越大, 对硝基电荷的影响就越大。通过对比发现, 当溶剂介电常数增大到一定程度时, 所有炸药分子硝基电荷的增加幅度都有所减小, 由于所有炸药分子在7种溶剂中的变化趋势大体一致, 所以, 可用其中一种炸药分子为例来讨论极性不同的7种溶剂对同一种炸药分子硝基电荷的影响, 仍以NQ为例, 图 5为其硝基电荷在7种溶剂中的变化曲线。其中0点对应的是气态分子的硝基电荷值, 可见, 介电常数大于丙酮(10.36)的溶剂, 其硝基电荷的变化值趋于平缓, 溶剂极性对它的影响逐渐达到饱和。
| 表 3 炸药分子在7种溶剂中的硝基电荷 Tab.3 Nitro group charges(e) of the explosive molecules in seven solvents |

|
图 4 7种溶剂中炸药分子硝基电荷变化趋势 Fig.4 Change trend of the nitro group charges of the explosive molecules in seven solvents |

|
图 5 NQ的硝基电荷在7种溶剂中的变化曲线 Fig.5 Change curve of the nitro group charges of NQ in seven solvents |
22种炸药分子在气态以及溶剂中的偶极矩值列于表 4。各炸药分子在溶剂中的偶极矩变化趋势如图 6所示。可见, 对于非极性炸药分子来说, 它们的偶极矩并不受溶剂极性的影响, 加入溶剂之后偶极矩仍然为0。对于绝大多数极性炸药分子而言, 它们的偶极矩会随着溶剂极性的增大而增大, 而且彼此间的变化值具有可比性, 所以, 仍然以NQ为例讨论不同溶剂对同种极性炸药分子的偶极矩的影响, 其在7种溶剂中的偶极矩变化如图 7所示。可见, 随着溶剂极性的增加, NQ的偶极矩呈增大趋势, 且介电常数大于丙酮的溶剂逐渐达到饱和。
| 表 4 炸药分子在7种溶剂中的偶极矩 Tab.4 Dipoles moments of the explosive molecules in seven solvents |

|
图 6 7种溶剂中炸药分子偶极矩变化趋势 Fig.6 Change trend of the dipole moments of the explosive molecules in seven solvents |

|
图 7 NQ的偶极矩在7种溶剂中的变化曲线 Fig.7 Change curve of the dipole moments of NQ in seven solvents |
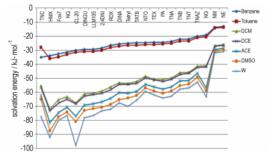
溶剂化能是衡量溶剂化效应大小的重要指标, 同时也可以在一定程度上反映溶质分子在溶剂中的稳定性, 溶剂化能的值越小, 绝对值越大, 则证明溶剂化效应越强烈, 相对的, 溶质的稳定性更高。22种炸药分子在各种溶剂中的溶剂化能列于表 5。图 8所示为22种炸药分子在各种溶剂中的溶剂化能变化趋势。可见, 随着溶剂极性的增加, 溶剂化能呈明显下降趋势, 且不同炸药分子变化趋势大体一致, 这里仍以NQ为例, 讨论不同极性的溶剂对某一种炸药分子的溶剂化能影响规律, 图 9为其溶剂化能在不同溶剂中的变化曲线。可见, 在丙酮之前, 随着极性的变化, 溶剂化能的变化比较明显, 在丙酮之后, 极性对溶剂化能的影响区域饱和, 极性的增加对溶剂化能的影响变小。
| 表 5 炸药分子在7种溶剂中的溶剂化能 Tab.5 Solvation energy of the explosive molecules in seven solvents |

|
图 8 炸药分子在7种溶剂中溶剂化能变化趋势 Fig.8 Change trend of the solvation energy of the explosive molecules in seven solvents |

|
图 9 NQ的溶剂化能在7种溶剂中的变化曲线 Fig.9 Change curve of the solvation energy of NQ in seven solvents |
键离解能可在一定程度上反映炸药分解难易程度, 从而衡量炸药的安全性。各炸药分子在不同溶剂中键离解能列于表 6, 对比发现, 对于不同类别的炸药, 溶剂对键离解能普遍存在微弱影响, 且规律并不明显, 除了两种小分子炸药NM和NE之外, 其余键离解能变化均只在4.2 kJ·
| 表 6 炸药分子在7种溶剂中的键离解能 Tab.6 Bond dissociation energy of the explosive molecules in seven solvents |
对比图 3、图 5、图 7、图 9发现,随着溶剂极性增大, NQ的键长、硝基电荷、极性以及溶剂化能的变化曲线形状几乎相同, 从以上讨论得知, 由于不同炸药在溶剂中上述参数变化趋势大体相同, 那么这种现象就可以推广到所计算的绝大多数炸药分子中。由于NQ的键长、硝基电荷、极性以及溶剂化能的变化曲线如此相近, 这就意味着它们之间存在内在联系, 由此可以推断溶剂化效应的整个过程:当炸药分子被放入溶剂中之后, 由于溶剂-溶质间的偶极-偶极相互作用, 使得溶质分子被极化, 体现为极性炸药分子偶极矩变大, 进而引起电荷的不平均分布更加明显, 体现为硝基电荷的变化, 这样由于电子结构变化又会引起几何结构变化, 体现为硝基所连接的特征键键长缩短, 这样又从结构改变引起能量变化, 这部分变化能量即为溶剂化能。
4 结论随着溶剂极性的增加, 绝大多数炸药分子的特征键键长、硝基电荷以及溶剂化能会有一定程度的减小, 偶极矩会体现为增加; 溶剂极性对炸药分子特征键键长、硝基电荷、偶极矩以及溶剂化能的影响是呈曲线形式的, 并以介电常数为10.36的丙酮为大概的分界线, 介电常数小于10.36, 极性的变化会对炸药分子产生较大的溶剂化效应的影响, 介电常数大于10.36时这种影响趋于平缓, 不随介电常数增加而增加; 溶剂化效应对绝大多数炸药分子的键离解能影响不大; 从不同溶剂对同种炸药分子的影响曲线上可以推断出溶剂化效应的影响过程:使溶质分子极化从而改变电子结构, 进而改变几何结构, 最终改变分子的能量, 产生溶剂化能。
| [1] |
Ignacy C, Krishna K G. A density functional theory-and atoms in molecules-based study of NINTA and NINTPA complexes toward physical properties controlling their stability[J].
Inorg Chem, 2010, 49: 6931-6941. DOI:10.1021/ic100453v |
| [2] |
Takumi H, Hideaki T, Masayoshi N, et al. QM/MM study combined with the theory of energy representation: Solvation free energies for anti/syn acetic acids in aqueous solution[J].
Chemical Physics Letters, 2006, 432: 240-244. DOI:10.1016/j.cplett.2006.10.079 |
| [3] |
Tomas Z, Zdenek C, Jaroslav V. Cisplatin interaction with cysteine and methionine in aqueous solution: computational DFT/PCM study[J].
J Phys Chem B, 2009, 113: 3139-3150. DOI:10.1021/jp807645x |
| [4] |
Paul R R. Computational analysis of the solvent wffect on the barrier to rotation about the conjugated C-N bond in methyl N, N-dimethylcarbamate[J].
J Org Chem, 2000, 65: 7930-7937. DOI:10.1021/jo000945z |
| [5] |
LI Ping, Bu Yu-xiang, AI Hong-qi. Density functional studies on conformational behaviors of glycinamide in solution[J].
J Phys Chem B, 2004, 108: 1405-1413. DOI:10.1021/jp036585b |
| [6] |
Kevin E R, Jiri V, Pavel H. Performance of the DFT-D method, paired with the PCM implicit solvation model, for the computation of interaction energies of solvated complexes of biological interest[J].
Phys Chem Chem Phys, 2007, 9: 5555-5560. DOI:10.1039/b708089a |
| [7] |
Bishwajit G, Dana A F, Marisa C K. Relevance of torsional effects to the conformational equilibria of 1, 5-diaza-cis-decalins: a theoretical and experimental study[J].
J Org Chem, 2001, 66: 1103-1108. DOI:10.1021/jo000580i |
| [8] |
Ikchoon L, Chang Kon Kim, In Suk Han. Theoretical studies of solvent effect on the basicity of substituted pyridines[J].
J Phys Chem B, 1999, 103: 7302-7307. DOI:10.1021/jp991115w |
| [9] |
Michael B, Nicolas S, Alain C. Water versus acetonitrile coordination to uranyl.density functional study of cooperative polarization effects in solution[J].
Inorg Chem, 2011, 50: 299-308. DOI:10.1021/ic101950d |
| [10] |
Mikael P and Peter A K. Ab Initio quantum mechanical and molecular dynamical study of intra-and intermolecular anhydride formation[J].
J Phys Chem A, 1999, 104: 8067-8074. |
| [11] |
CHEN Xue-nian, BAO Xiao-guang, ZHAO Ji-cheng, et al. Experimental and computational study of the formation mechanism of the diammoniate of diborane: the role of dhydrogen bonds[J].
J Am Chem Soc, 2011, 133: 14172-14175. DOI:10.1021/ja203648w |
| [12] |
ZHANG Chao-yang, LI Yu-zhen, XIONG Ying, et al. Acid and alkali effects on the decomposition of HMX molecule: a computational study[J].
J Phys Chem A, 2011, 115: 11971-11978. |
| [13] |
柴涛, 周圣, 张景林. 超临界流体反溶剂法制备高聚物粘结炸药技术初探[J].
含能材料, 2005, 13(4): 205-207. CHAI Tao, ZHOU Sheng, ZHANG Jing-lin. Preparation of PBX by supercritical fluid anti-solvent method[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2005, 13(4): 205-207. |
| [14] |
冯岩岩. 炸药生产中常用有机溶剂回收的试验研究[D]. 南京理工大学. 2012.
|
| [15] |
王保民, 张景林. 气体反溶剂(GAS)过程细化技术及对炸药安全性能的影响研究[J].
中国安全科学学报, 2001, 11(4): 32-34. WANG Bao-min, ZHANG Jing-lin. Study on fining technology of gas anti-solvent (GAS) process and its effect on safety of the explosive[J]. China Safety Science Journal, 2001, 11(4): 32-34. |
| [16] |
Melius C F. The thermochemistry and reaction pathways of energetic material decomposition and combustion[J].
Philosophical Transactions: Physical Sciences and Engineering, 1992, 339: 365-376. DOI:10.1098/rsta.1992.0042 |
| [17] |
Kiselev V G, Gritsan N P. Theoretical study of the primary processes in the thermal decomposition of hydrazinium nitroformate[J].
J Chem Phys A, 2009, 113: 11067-11074. DOI:10.1021/jp906853e |
| [18] |
陈锋. 乳化炸药与有机溶剂混合物热分解机理研究[D]. 安徽: 安徽理工大学, 2006.
|
| [19] |
姬广富. 高能钝感炸药分子和晶体的结构和性能的理论研究[D]. 南京: 南京理工大学, 2002.
|
| [20] |
Becke, A. D.. Density-functional thermochemistry. Ⅲ. the role of exact exchange[J].
J Chem Phys, 1993, 98: 5648-5652. DOI:10.1063/1.464913 |
| [21] |
Lee C, Yang W, Parr R G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].
Phys Rev B, 1988, 37: 785 DOI:10.1103/PhysRevB.37.785 |
| [22] |
Eckert F K. Fast solvent screening via quantum chemistry: COSMO-RS approach[J].
AIChE J, 2002, 48: 369-385. DOI:10.1002/(ISSN)1547-5905 |
| [23] |
Frisch M. J, Trucks G W, Schlegel, H B, et al. Gaussian03[CP], Revision B.05; Gaussian[CP], Inc: Pittsburgh, PA, 2003.
|
| [24] |
ZHANG Chao-yang, SHU Yuan-jie, HUANG Yi-gang, et al. Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds[J].
J Phys Chem B, 2005, 109: 8978-8982. DOI:10.1021/jp0512309 |
The effects of seven solvents on the feature bond lengths, nitro group charges, dipole moment, solvation energy and bond dissociation energy of 22 common explosives were investigated based on DFT and PCM.