




2. 国家民用爆破器材质量监督检测中心,江苏 南京 210094
2. National Civil Blasting Equipment Quality Supervision and Testing Center, Nanjing 210094, China
有机叠氮化合物是一类广受关注的含能物质, 具有独特的作用和性能, 可用作含能粘合剂、含能增塑剂、含能氧化剂等[1-6]。然而, 有机叠氮化合物普遍存在密度小的问题。众所周知, 密度是评估高能化合物性能的一个重要的参数, 其大小影响并决定着化合物的爆轰性能, 故如何提高密度一直是含能物质研究中的热门话题。季戊四醇的衍生物——四叠氮甲基丁烷(TAPE), 密度仅为1.444 g·
运用Gaussian 03[16]程序包中的B3LYP[17-19]方法和6-31

|
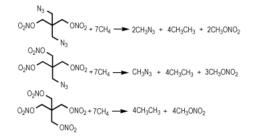
Scheme 1 |
在此基础上, 运用Kamlet-Jacobs (K-J)公式[21]计算爆速和爆压:
| $\begin{eqnarray} D=1.01 (N \bar{M}^{1/2} Q^{1/2} )^{1/2} (1 + 1.30ρ) \end{eqnarray}$ | (1) |
| $\begin{eqnarray} p=1.558ρ^{2} N \bar{M}^{1/2} Q^{1/2} \end{eqnarray}$ | (2) |
式中,
采用与以往研究相同的方法[22-24]由(3)式计算分子中较弱键的键离解能(
| $\begin{eqnarray} E_{\text{BD}} (A-B)=E(A·)+E(B·)-E(A-B) \end{eqnarray}$ | (3) |
式中,A·和B·为A-B键解离产物,

|
由Peter Politzer等[25]提出的方法求比冲
| $\begin{eqnarray} I_{\text{s}}=T_{\text{C}}~^{1/2}N^{1/2 } \end{eqnarray}$ | (4) |
| $\begin{eqnarray} \text{Δ}H_{\text{C}}=C_{\text{p},\text{g}}(T_{\text{C}} -T_{0}) \end{eqnarray}$ | (5) |
| $\begin{eqnarray} \text{Δ}H_{\text{C}}=∑\varDelta H_{\text{f},\text{R}}-∑\varDelta \text{H}_{f,P} \end{eqnarray}$ | (6) |
式中,
图 1给出了标题物优化后的几何构型以及部分键的键长和Mulliken集居数。Mulliken集居数[26]分析尽管有不足, 但对于反映系列分子中电子分布的变化趋势仍然是有意义的。一般而言, Mulliken键集居数越小, 表明该键越弱。从图 1可以看出, 对于分子中只含有叠氮基的TAPE而言, C—N键集居数(0.210)比—

|
图 1 标题物的几何结构(图中数据分别表示键长(上)和Mulliken集居数(下)) Fig.1 Optimized geometric configuration of the title compounds. The data are bond length and Mulliken bond population from the top to the bottom, respectively |
根据统计热力学原理[27], 由优化所得结构和采用因子0.96校正后的频率[28-29], 计算了标题物在200~800 K温度范围的热力学函数, 所得结果列于表 1,热力学函数与温度的关系见图 2, 由图 2拟合得到的定量关系式见表 2。
|
表 1 标题物在200~800 K时的标准摩尔热容、标准摩尔熵和标准摩尔焓
Tab.1 Standard molar heat capacity ( |

|
图 2 热力学函数与温度的关系
Fig.2 Relationships between |
|
表 2 热力学函数与温度的回归关系式
Tab.2 Regression equations between |
由表 1和图 2可见, 标准摩尔热容(
计算所得HOF(

|
图 3 生成热 |
计算得到太安的
表 3给出了标题物的
|
表 3 密度、爆速、爆压、氧平衡和比冲
Tab.3 Density ( |

|
图 4 密度、爆热、爆速、爆压、氧平衡和比冲与叠氮基数目 |
根据键集居数结果, O—
|
表 4 前线分子轨道能级差、键解离能和氢转移反应活化能
Tab.4 Energy gaps ( |
一般来说,

|
图 5 TAPE的氢转移反应过渡态的优化结构
Fig.5 Optimized structure of the transition state for the |
(1) 叠氮基对生成热的贡献大于硝酸酯基, 而硝酸酯基对爆轰性能的贡献比叠氮基大。
(2) 对于取代基只有—
(3) PATN的稳定性和爆轰性能均接近于PETN, 且优于PDADN, 是一种有潜在研究价值的高能化合物。
| [1] |
施明达. 一种新型含能材料——叠氮有机化合物[J].
火炸药, 1992(4): 24-30. SHI Ming-da. A new kind of energetic material——organic azido compounds[J]. Chinese Journal of Explosives Propellants, 1992(4): 24-30. |
| [2] |
Provatas A. Energetic polymers and plasticizers for explosive formulations——A review of recent advances[R]. DSTO-TR-0966, 2000.
|
| [3] |
Mohan Y M, Mani Y, Raju K M. Synthesis of azido polymers as potential energetic propellant binders[J].
Designed Monomers and Polymers, 2006, 9(3): 201-236. DOI:10.1163/156855506777351045 |
| [4] |
Klapötke T M, Krumm B, Steemann F X. Preparation, characterization, and sensitivity data of some azidomethyl nitramines[J].
Propellants Explosives Pyrotechnics, 2009, 34(1): 13-23. DOI:10.1002/prep.v34:1 |
| [5] |
Mohan Y M, Raju K M, Sreedhar B. Synthesis and characterization of glycidyl azide polymer with enhanced azide content[J].
Internationa Journal of Polymeric Materials, 2006, 55(6): 441-455. DOI:10.1080/009140390970486 |
| [6] |
Kumari D, Balakshe R, Banerjee S, et al. Energetic plasticizers for gun rocket propellants[J].
Review Journal of Chemistry, 2012, 2(3): 240-262. DOI:10.1134/S207997801203003X |
| [7] |
欧育湘.
炸药学[M]. 北京: 北京理工大学出版社, 2006.
OU Yu-xiang. Explosives[M]. Beijing: Beijing Institute of Technology Press, 2006 |
| [8] |
王平, 李顺秀, 黄悦, 等. 二叠氮季戊二醇二硝酸酯的研究[J].
含能材料, 1994, 2(3): 29-35. WANG Ping, LI Shun-xiu, HUANG Yue, et al. Investigation of pentaerythritol diazido dinitrate[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 1994, 2(3): 29-35. |
| [9] |
王进, 李疏芬, 阴翠梅, 等. 含能增塑剂PDADN合成及性能研究[J].
固体火箭技术, 1999, 22(3): 41-45. WANG Jin, LI Shu-fen, YIN Cui-mei, et al. Study on synthesis and properties of energetic plasticizer PDADN[J]. Journal of Solid Rocket Technology, 1999, 22(3): 41-45. |
| [10] |
王进, 李疏芬, 张晓宏, 等. 叠氮硝酸酯对硝胺改性双基推进剂燃烧性能的影响[J].
火炸药学报, 2001(2): 22-31. WANG Jin, LI Shu-fen, ZHANG Xiao-hong, et al. Effect of azide nitrate ester oil combustion behavior of nitramine modified double base propellant[J]. Chinese Journal of Explosives Propellants, 2001(2): 22-31. |
| [11] |
苏天铎, 张丽洁, 柳恒, 等. 二叠氮季戊二醇二硝酸酯合成新工艺[J].
火炸药学报, 2005, 28(1): 52-60. SU Tian-duo, ZHANG Li-jie, LIU Heng, et al. A new technique for preparation of pentaerythritol diazido dinitrate[J]. Chinese Journal of Explosives Propellants, 2005, 28(1): 52-60. |
| [12] |
王遵尧, 贡雪东, 肖鹤鸣. 二叠氮季戊二醇二硝酸酯的AM1分子轨道研究[J].
含能材料, 1996, 4(4): 157-162. WANG Zun-yao, GONG Xue-dong, XIAO He-ming. AM1 molecular orbital study of pentaerythritol diazido dinitrate[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 1996, 4(4): 157-162. |
| [13] |
王遵尧, 肖鹤鸣. 二叠氮季戊二醇二硝酸酯的构象和IR谱的密度泛函理论研究[J].
含能材料, 2000, 8(2): 63-66. WANG Zun-yao, XIAO He-ming. A Study on the conformations and IR spectra of pentaerythritol diazido dinitrate by density functional theory[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2000, 8(2): 63-66. |
| [14] |
Feng L M, Yang Y, Zhang L J, et al. Synthesis and characterization of pentaerythritol triazide mononitrate (PTAMN)[C]∥35th International Annual Conference of ICT. Berghausen; Fraunhofer-Institut fur Chemische Technologie. 2004:145.1-145.4.
|
| [15] |
Quinto-Hernandez, Alfredo, Wodtke A M, Bennett C J, et al. On the interaction of methyl azide (CH3N3) ices with ionizing padiation: formation of methanimine (CH2NH), hydrogen cyanide (HCN), and hydrogen isocyanide (HNC)[J].
The Journal of Physical Chemistry A, 2010, 115(3): 250-264. |
| [16] |
Frisch M, Trucks G, Schlegel H, et al. Gaussian 03[CP], revision B. 03. Gaussian Inc. Pittsburgh, PA. 2003.
|
| [17] |
Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].
Physical Review B, 1988, 37(2): 785-789. DOI:10.1103/PhysRevB.37.785 |
| [18] |
Becke A D. Density-functional thermochemistry. Ⅱ. The effect of the Perdew-Wang generalized-gradient correlation correction[J].
The Journal of Chemical Physics, 1992, 97(12): 9173-9177. DOI:10.1063/1.463343 |
| [19] |
Becke A D. Density-functional thermochemistry. Ⅲ. The role of exact exchange[J].
The Journal of Chemical Physics, 1993, 98(7): 5648-5652. DOI:10.1063/1.464913 |
| [20] |
Hariharan P C, Pople J A. The influence of polarization functions on molecular orbital hydrogenation energies[J].
Theoretica Chimica Acta, 1973, 28(3): 213-222. DOI:10.1007/BF00533485 |
| [21] |
Kamlet M J, Jacobs S. A simple method for calculating detonation properties of C—H—N—O explosives[J].
The Journal of Chemical Physics, 1968, 48: 23-35. DOI:10.1063/1.1667908 |
| [22] |
Wang G X, Gong X D, Xiao H M. Theoretical investigation on density, detonation properties, and pyrolysis mechanism of nitro derivatives of benzene and aminobenzenes[J].
International Journal of Quantum Chemistry, 2009, 109(7): 1522-1530. DOI:10.1002/qua.v109:7 |
| [23] |
Zhang J Y, Du H C, Wang F, et al. DFT studies on a high energy density cage compound 4-Trinitroethyl-2, 6, 8, 10, 12-pentanitrohezaazaisowurtzitane[J].
The Journal of Physical Chemistry A, 2011, 115(24): 6617-6621. DOI:10.1021/jp1118822 |
| [24] |
Wang F, Du H C, Zhang J Y, et al. Computational studies on the crystal structure, thermodynamic properties, detonation performance, and pyrolysis mechanism of 2, 4, 6, 8-Tetranitro-1, 3, 5, 7-tetraazacubane as a novel high energy density material[J].
The Journal of Physical Chemistry A, 2011, 115(42): 11788-11795. DOI:10.1021/jp2049469 |
| [25] |
Politzer P, Murray J, Grice M, et al.
Chemistry of energetic materials[M]. New York: Academic Press, San Diego, CA, 1991 |
| [26] |
Mulliken R S. Electronic population analysis on LCAO [Single Bond] MO molecular wave functions[J].
The Journal of Chemical Physics, 1955, 23(10): 1833-1840. DOI:10.1063/1.1740588 |
| [27] |
Hill T L.
An introduction to statistical thermodynamics[M]. New York: Courier Dover Publications, 1960 |
| [28] |
Scott A P, Radom L. Harmonic vibrational frequencies: an evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors[J].
The Journal of Physical Chemistry, 1996, 100(40): 16502-16513. |
| [29] |
Wong M W. Vibrational frequency prediction using density functional theory[J].
Chemical Physics Letters, 1996, 256(4): 391-399. |
| [30] |
金韶华, 李文, 松全才. 有机叠氮化物的热分解[J].
含能材料, 1998, 6(3): 123-127. JIN Shao-hua, LI Wen, SONG Quan-cai. The thermal decomposition of organic azide[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 1998, 6(3): 123-127. |
| [31] |
杜洪臣, 许晓娟, 刘彦, 等. 苯的硝基和叠氮基衍生物的理论研究[J].
化学学报, 2011, 69(3): 269-276. DU Hong-chen, XU Xiao-juan, LIU Yan, et al. Theoretical studies on the nitro and azido derivatives of benzene[J]. Acta Chimica Sinica, 2011, 69(3): 269-276. |
| [32] |
Liu Y, Gong X D, Wang L J, et al. Substituent effects on the properties related to detonation performance and sensitivity for 2, 2′, 4, 4′, 6, 6′-hexanitroazobenzene derivatives[J].
The Journal of Physical Chemistry A, 2011, 115: 1754-1762. DOI:10.1021/jp200512j |
| [33] |
Xu X J, Zhu W H, Gong X D, et al. Theoretical studies on new potential high energy density compounds (HEDCs) adamantyl nitrates from gas to solid[J].
Science in China Series B: Chemistry, 2008, 51(5): 1-13. |
| [34] |
Chen Z X, Xiao J M, Xiao H M, Chiu Y N. Studies on heats of formation for tetrazole derivatives with density functional theory B3LYP method[J].
The Journal of Physical Chemistry A, 1999, 103(40): 8062-8066. DOI:10.1021/jp9903209 |
| [35] |
Gong X D, Xiao H M, Tian H. Comparative studies on the structures, infrared spectrum, and thermodynamic properties of phthalocyanine using ab initio Hartree-Fock and density functional theory methods[J].
International Journal of Quantum Chemistry, 2002, 86(6): 531-540. DOI:10.1002/(ISSN)1097-461X |
| [36] |
Jursic B. Computing the heat of formation for cubane and tetrahedrane with density functional theory and complete basis set ab initio methods[J].
Journal of Molecular Structure: theochem, 2000, 499(1): 137-140. |
| [37] |
肖鹤鸣, 许晓娟, 邱玲.
高能量密度材料的理论设计[M]. 北京: 科学出版社, 2008.
XIAO He-ming, XU Xiao-juan, QIU Ling. Theoretical Design of High Energy Density Materials[M]. Beijing: Science Press, 2008 |
A systematic theoretical study was carried out on tetraazido pentaerythritol (TAPE), pentaerythritol triazido nitrate (PTAN), pentaerythritol diazido dinitrate (PDADN), pentaerythritol azido trinitrate (PATN) and pentaerythritol tetranitrate (PETN) to investigate their structures and properties, especially the pyrolysis mechanism by analyzing the bond dissociation energy (EBD) of the possible trigger bond and the activation energy (Ea) of the hydrogen transfer reaction.