




2. 北京理工大学 化学学院, 北京 100081
2. School of Chemistry, Beijing Institute of Technology, Beijing 100081, China
目前, 含能材料的研究已进入高能量密度材料(HEDM)的新阶段[1]。作为高氮含能化合物, 氨基四唑类含能化合物在高能钝感低毒炸药[2]、高产气量气体发生剂[3-4]、环保型烟火药[5-6]、低特征信号固体推进剂添加剂[7-8]等领域有广阔的应用前景。这类化合物是当前高能量密度含能材料的研究热点之一。
氨基四唑主要有两种, 即5-氨基四唑(5-AT)和1, 5-二氨基四唑(DAT), 这两种化合物均是制备其它富氮化合物的重要中间体。1, 5-二氨基四唑(DAT)具有很高的氮含量(84%)、正生成焓及很好的热稳定性[9], 可与强酸反应得到1, 5-二氨基四唑盐酸盐、硝酸盐和高氯酸盐[10]。经季铵化反应后, 还可用来制备其硝酸盐、二硝基氨盐及叠氮盐[11]。DAT的阳离子也可与有机杂环阴离子成盐[12]。另外, DAT分子中每个N原子上都有一对孤对电子, 都有可能成为配位原子, 是一种以N原子为配位原子的多齿配体。以DAT为配体, Cu、Mn、Co、Zn、Cd等作中心离子,
关于DAT合成的报道并不多, 1933年, Stollé等[20]合成出DAT; Gaponik等[21]以氨基硫脲、氧化铅和叠氮化钠为原料制备出DAT, Pavel等[22]对其进行了改进; 直到Gálvez-Ruiz等[23]以二氨基胍盐酸盐为原料制备DAT, 产率达到58%。
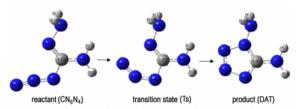
本研究基于密度泛函理论, 对1, 5-二氨基四唑(DAT)进行电子结构性质分析,并对Gálvez-Ruiz合成DAT方法中关键的成环反应(图 1)机理和动力学进行研究, 以获得其异构化反应机理, 和气相状态下成环反应动力学常数, 为氨基四唑类高氮含能材料前驱体的实验研究提供理论基础。

|
图 1 叠氮化物异构化合成1, 5-二氨基四唑(DAT)反应 Fig.1 The isomerization reaction of azide for1, 5-diamino-tetrazole (DAT) |
基于密度泛函理论, 对于1, 5-二氨基四唑及叠氮化物异构化合成DAT反应涉及的所有驻点(包括反应物、过渡态和产物)的几何结构、振动频率、自然键轨道、以及零点能(ZPE)的计算均是在B3LYP/6-311G水平下完成的。为确认反应过渡态(TS)是否连接反应物和产物两个极小值点, 进一步运用内禀反应坐标理论(IRC)对过渡态进行计算。在IRC计算中设定步长为0. 02(amu)
对叠氮化物的成环异构化反应动力学的考察, 是以上述计算为基础的。通过VKLab[24]和POLYRATE 8. 2程序包[25], 采用了包括传统过渡态理论(TST)、Eckart隧道校正理论(TST/ Eckart)[26], 以及变分过渡态理论(CVT)[27-29], 计算得到200~1000 K区间内的反应速率常数。所有计算均是通过Gaussian03程序包[30]计算完成的。
3 结果与讨论 3.1在B3LYP/ 6-311G水平下优化得到1, 5-二氨基四唑(DAT)的几何结构如图 2所示。表 1列出了其优化结构的部分几何参数。

|
图 2 B3LYP/ 6-311G水平下1, 5-二氨基四唑(DAT)的几何优化结构 Fig.2 Optimized geometry structure for 1, 5-diamino-tetrazole (DAT) with B3LYP/6-311G |
| 表 1 B3LYP/6-311G几何结构计算中DAT部分键长和角度 Tab.1 Selected bond lengths and angles of the optimized geometry for DAT by B3LYP/6-311G calculations |
由图 2可知, 1, 5-二氨基四唑(DAT)的分子结构不具有对称性, 所有N原子与C原子均在同一平面, 形成共平面的五元环。从表 1数据键长来看, 四唑环上化学键由于
在B3LYP/ 6-311G水平下优化得到1, 5-二氨基四唑(DAT)的几何结构的基础上, 对DAT分子进行自然键轨道(NBO)分析, 其NBO电荷分布见表 2。从表 2的NBO电荷分布来看, DAT分子中由于N原子的吸电子性质, N原子均带负电荷, 从而使得C原子显示出较高的正电性。其中四唑环上的N(4), N(5)所带电荷分别为: -0.377和-0.126a.u., 这是由于—
| 表 2 1, 5-二氨基四唑(DAT)的NBO电荷 Tab.2 NBO charges of 1, 5-diamino-tetrazole (DAT) |
采用密度泛函理论, 在B3LYP/6-311G水平下对反应物、产物和过渡态分别进行了几何结构优化及振频率分析。图 3为叠氮化物关环合成DAT反应历程,反应中所涉及的驻点优化后得到的部分几何参数见表 3。由表 3可知, 从反应物、过渡态到产物DAT, 观察其键长、键角变化情况, 反应物主体结构变化很小, 主要是C(1)原子上所连的叠氮基在反应过程中逐渐关环。N(2)—N(3)—N(4)键长均有不同程度增加, 末端N(2)原子逐渐与电负性较强的N(5)原子成环, 且环上所涉及的分子键长逐渐平均化, 均在1.30~1.40 nm之间; 分子中所有C原子和N原子均处在同一个平面, 且键长分布均匀, 键角趋于108°左右, 说明分子共轭性较大。整个过程叠氮基关环趋势明显, 分子趋于稳定五元环结构。

|
图 3 叠氮化物的关环合成DAT反应历程 Fig.3 Cycling reaction of azide for1, 5-diamino-tetrazole (DAT) |
| 表 3 B3LYP/6-311G水平下叠氮化物异构化反应中的各驻点部分优化几何参数 Tab.3 The optimized geometric parameters of the reactant, transition state and product for azide isomerization at B3LYP/6-311G level of theory |
计算DAT异构化反应在B3LYP/6-311G水平下得到的能量参数, 包括总能量变化(
在B3LYP/6-311G水平下进行IRC计算, 结果表明过渡态的确连接反应物和产物。在相同基组条件下, 采用CCSD(T)方法进一步优化最小能量路径(MEP)上各点的能量。图 4描述了反应的振动绝热基态势

|
图 4 叠氮化物的关环合成DAT反应势能曲线 Fig.4 Potential energies for isomerization reaction of azide for DAT |
产物DAT相对反应物能量较低, 因而产物分子结构更加稳定; 比较正反应和逆反应的能垒大小, 正反应相对更容易进行, 具有热力学可行性; 另外, 经零点能校正的振动绝热基态势要比经典位垒的数值更加准确可靠, 该势能曲线很好地反映了叠氮基关环形成DAT的能量变化过程。
3.2.3 速率常数的计算异构化合成DAT反应的速率常数均是在B3LYP/6-311G水平下所得最小能量途径MEP基础上, 通过TST、TST/Eckart和CVT(

|
图 5 叠氮化物关环合成DAT反应速率的Arrhenius曲线 Fig.5 The Arrhenius curves for the isomerization reaction of azide for DAT |
反应速率常数随温度变化关系满足Arrhenius经验公式, 由于温度范围较宽, 活化能与温度有关, 采用修正的三参量方程式加以校正。根据不同方法的计算结果, 拟合得到如下速率常数:
| $\begin{eqnarray} &&k(T)=1.52×10^{12}×T^{-0.01278}×\text{e}^{-(8.54×10^{3}/T)}s^{-1}(\text{TST})\\ &&k(T)=1.31×10^{12}×T^{0.00554}×\text{e}^{-(8.47×10^{3}/T)}s^{-1}(\text{TST}/\text{Eckart})\\ &&k(T)=7.11×10^{14}×T^{-0.01278}×\text{e}^{-(9.61×10^{3}/T)}s^{-1}(\text{CVT}) \end{eqnarray}$ |
本文基于密度泛函理论, 在B3LYP/6-311G水平下对DAT的电子结构性质及异构化反应动力学进行研究, 得到如下结论:
(1) 1, 5-二氨基四唑(DAT)的分子结构不具有对称性, 所有N原子与C原子均在同一平面, 形成稳定的四唑环。从NBO电荷分布来看, DAT分子中四唑环上的N(4), N(6), N(9)相对负电荷最多, 但由于N(6)与四唑环存在共轭, 因而DAT分子中的N(4)和N(9)易参与金属原子配位, 形成一系列以DAT为配体的配位化合物。
(2) 相同基组和方法下对叠氮化物异构化合成DAT的关环反应的过渡态进行了结构优化和频率分析。采用内禀反应坐标的方法, 获得最小反应能量路径及势能曲线。
(3) 计算结果表明该反应为放热反应, 自发正方向进行, 反应活化能较低; 并通过TST、TST/Eckart和CVT(
| [1] |
周世光, 吴文健. 高能量密度材料[J].
化工时刊, 1997, 11(12): 3-6. ZHOU Shi-guang, WU Wen-jian. High energy density materials[J]. Chinese Industry Times, 1997, 11(12): 3-6. |
| [2] |
Klapötke T M. New nitrogen-rich high explosives[J].
Stucture and Bonding, 2007, 125: 85-121. DOI:10.1007/978-3-540-72202-1 |
| [3] |
Hiskey M, Chavez D, Naud D L, et al. Low smoke pyrotechnic compositions: US 6312537 [P]. 2001.
|
| [4] |
Khandhadia P S, Burns S P. Thermally stable nonazide auto motive airbag propellants: US 6306232[P]. 2001.
|
| [5] |
Ali A N, Son S F, Hiskey M, et al. Novel high-nitrogen propellant use in solid fuel micropropulsion[J].
Journal of Propulsion and Power, 2004, 20: 120-126. DOI:10.2514/1.9238 |
| [6] |
Chavez D, Hiskey M, Naud D L. High-nitrogen fuels for low-smoke pyrotechnics[J].
Journal of Pyrotechnics, 1999, 10: 17-36. |
| [7] |
Miller C G, Williams G K. Gas generant compositions: US 0199937[P]. 2009.
|
| [8] |
Miller C G, Williams G K. Water-based synthesis of poly (tetrazoles):US 0099111[P]. 2008.
|
| [9] |
Kozyro A A S, Krasulin A P, Sevruk V M, et al. Thermodynamic properties of tetrazole derivatives in different aggregation states[J].
Zhurnal Fizicheskoi Khimii, 1990, 64(3): 656-661. |
| [10] |
Ra ap, R. Amination of tetrazoles with hydroxylamine-O-sulfonic acid: 1-and 2-aminotetrazoles[J].
Canadian Journal of Chemistrt-Revue Canadian De Chimie, 1969, 47(19): 3677-3681. DOI:10.1139/v69-606 |
| [11] |
Gálvez-Ruiz J C, Holl G, Karaghiosoff K, et al. Derivatives of 1, 5-diamino-1H-tetrazole: A new family of energetic heterocyclicbased salts[J].
Inorganic Chemistry, 2005, 44(12): 4237-4253. DOI:10.1021/ic050104g |
| [12] |
Tao G H, Guo Y, Parrish D A, et al. Energetic 1, 5-diamino-4H-tetrazolium nitro-substiuted azolates[J].
Materials Chemistry, 2010, 20: 2999-3005. DOI:10.1039/b925267c |
| [13] |
Smirnov A V, Ilyushin M A, Tselinskii Ⅳ. Synthesis of cobalt (Ⅲ) ammine complexes as explosives for safe priming charges[J].
Russian Journal of Applied Chemistry, 2001, 77(1): 99-102. |
| [14] |
臧艳. 1, 5-二氨基四唑及其含能化合物研究(Ⅰ)[D]. 北京: 北京理工大学, 2008.
|
| [15] |
崔燕. 1, 5-二氨基四唑和3-叠氮-1, 2, 4-三唑含能配合物研[D]. 北京: 北京理工大学, 2008.
|
| [16] |
Gaponik P N, Voitekhovich S V, Lyakhov A S, et al. Crystal structure and physical properties of the new 2D polymeric compound bis (1, 5-diaminotetrazole) dichlorocopper (Ⅱ)[J].
Inorganica Chimica Acta, 2005(8): 2549-2557. |
| [17] |
齐书元, 张同来, 杨利, 等. 1, 5-二氨基四唑及其系列化合物研究进展[J].
含能材料, 2009, 17(4): 486-489. QI Shu-yuan, ZHANG Tong-lai, YANG Li, et al. Progress in 1, 5-diamino-1H -tetrazole and its derivatives[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(4): 486-489. |
| [18] |
Zhang J G, Li Z M, Zang Y, et al. Synthesis, structural investigation and thermal properties of a novel manganese complex Mn2(DAT)2Cl4(H2O)4(DAT=1, 5-diaminotetrazole)[J].
Journal of Hazardous Materials, 2010, 178(1-3): 1094-1099. DOI:10.1016/j.jhazmat.2010.02.063 |
| [19] |
Li Z M, Zhang J G, Zang Y, et al. A novel nitrogen-rich cadmium coordination compound based on 1, 5-diaminotetrazole: Synthesis, structure investigation, and thermal properties[J].
Journal of Chemical & Engineering Data, 2010, 55(9): 3109-3116. |
| [20] |
Stollé R, Netz H, Kramer O, et al. Derivatives of 1-aminotetrazoles[J].
Journal of Fur Praktische Chemie-Practical Applications and Chemistry, 1933, 138: 1-17. DOI:10.1002/prac.19331380101 |
| [21] |
Gaponik P N, KaravaiV P. Sythesis and properties of 1, 5-diaminotetrazole[J].
Khim Geterotsikl Soedin, 1984, 12: 1683-1686. |
| [22] |
Pavel N Gaponik, Sergei V Voitekhovich, Alexander S Lyakhov. Crystal structure and physical properties of the new 2D polymeric compound bis(1, 5-diaminotetrazole) dichlorocopper(Ⅱ)[J].
Inorganica Chimica Acta, 2005, 358: 2549-2557. DOI:10.1016/j.ica.2005.03.005 |
| [23] |
Gálvez-Ruiz J C, Holl G, Karaghiosoff K, et al. Derivatives of 1, 5-diamino-1H-tetrazole: A new family of energetic heterocyclicbased salts[J].
Inorganic Chemistry, 2005, 44(12): 4237-4253. DOI:10.1021/ic050104g |
| [24] |
ZHANG Shao-wen, Truong T N. VKLab version1.0[CP/ CD]. Minneapolis: University of Minnesota, Utah: University of Utah, 2001.
|
| [25] |
Chuang Y Y, Corchado J C, Fast P L. POLYRATE, Program vision 8.2 Minneapolis[CP/CD]. Minneapolis: University of Minnesota, 1999.
|
| [26] |
Truong N T, Truhar D G. Ab initio transition state theory calculation of the raction rate for OH+CH4→H2O+CH3[J].
Journal of Chemical Physics, 1990, 93: 1761-1769. DOI:10.1063/1.459103 |
| [27] |
Miller W H. Tunneling corrections to unimolecular rate constants, with application to formaldehyde[J].
Journal of American Chemical Society, 1979, 101(23): 6810-6814. DOI:10.1021/ja00517a004 |
| [28] |
Truhlar D G, Garrett B C. Variational transition state theory[J].
Annual Review of Physical Chemistry, 1984, 35: 159-189. DOI:10.1146/annurev.pc.35.100184.001111 |
| [29] |
Truong N T. A direct ab initio dynamics approach for calculating thermal rate constants using variational transition state theory and multidimensional semiclassical tunneling methods[J].
Journal of Chemical Physics, 1994, 100(11): 8014-8025. DOI:10.1063/1.466795 |
| [30] |
Frisch M J, Trucks G W, Schlegel H B. Gaussian 03.Revision A. 01 [CP/ CD]. Pittsbur g h PA: Gaussian Inc, 2003.
|
The electronic properties of 1, 5-diamino-tetrazole(DAT) and isomerization reaction kinetics were studied based on density functional theory.