




2. 中国工程物理研究院流体物理研究所, 四川 绵阳 621900;
3. 嘉兴学院生物与化学工程学院, 浙江 嘉兴 314001
2. Institute of Fluid Physics, China Academy of Engineering Physics, Mianyang 621900, China;
3. College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, China
奥克托今(HMX, C4H8O8N8)是当前综合性能最好的单体炸药, 已作为重要组分广泛应用于混合炸药和固体推进剂等复合含能材料中。国内外已有较多关于HMX晶体[1-6]和以HMX为基混合炸药如高聚物粘结炸药(PBX)[7-14]等的分子动力学(MD)模拟研究。但其中专门讨论建立模型的文献很少。肖继军等[7, 15, 16]在PBXs的MD模拟中, 曾经建议过“吸附包覆”、“渗透添加”和“切割分面”三种模型, 主要用于研究TATB和HMX等基炸药与氟聚物之间的结合能和力学性能。而对于基炸药晶体模型的讨论, 特别是其模型选取对涉及安全性(如感度)判别的MD模拟结果的影响问题, 至今尚未见有报道。因为力场选定之后, 影响MD模拟结果的关键因素就是模型建立, 故本工作选择HMX晶体为研究对象, 通过MD模拟, 对其超晶胞的大小和形状以及是否切割分面和切割深度等模型建立问题, 作细致地比较研究。从求得的结构(如引发键的键长分布)、相互作用能(如引发键连双原子作用能)和力学性能及其对感度的关联等方面, 提供模型对MD模拟结果影响的基础信息, 希望可对后续高能材料结构和性能(特别是安全性和力学性能)的MD模拟工作有所助益。
2 模型搭建和模拟细节 2.1 模型搭建以β -HMX中子衍射获得的晶体数据[17]为依据, 搭建HMX晶胞模型[18]。选取(4×2×4)超晶胞, 含64个HMX分子, 共1792个原子, 由此得到的纯HMX周期箱记为模型Ⅰ; 对(4×2×4)超晶胞沿(1 0 0)晶面方向进行“切割”, 并使Z轴平行晶轴c向量, 同时垂直于(1 0 0)晶面, 晶面(1 0 0)面积为2.21×3.48 nm2, c方向上取深度1即OC=2.38 nm其上真空层高度设置为0, 这样得到的晶体模拟周期箱, 记作模型Ⅱ(原子数亦为1792)。用同样的方法在c方向取深度1.5即OC=3.46 nm, 亦得到另一种晶体模拟周期箱, 记作模型Ⅲ(原子数为2688)。模型Ⅰ、Ⅱ和Ⅲ的初始模型如图 1所示。

|
图 1 (4×2×4)HMX超晶胞的Ⅰ、Ⅱ和Ⅲ初始模型 Fig.1 Initial structure models of Ⅰ, Ⅱ and Ⅲ for HMX crystal |
类似地, 选取(4×4×4)超晶胞, 含128个HMX分子, 共3584个原子, 该纯HMX周期箱记为模型Ⅰ′。仿照(4×2×4)超晶胞“切割”模型的方法, 对(4×4×4)超晶胞进行“切割”, 此情况下晶面(1 0 0)面积为4.42×3.48 nm2, 同样在c方向上分别取深度1即OC=2.38 nm和深度1.5即OC=3.46nm得到另两种模型, 分别记为模型Ⅱ′(原子数3584)和模型Ⅲ′(原子数5376), 为节省篇幅略去了模型Ⅰ′、Ⅱ′和Ⅲ′的初始模型图示。
2.2 计算方法和细节将搭建好的β -HMXⅠ、Ⅱ、Ⅲ, Ⅰ′、Ⅱ′和Ⅲ′共6种超胞模型, 利用Materials Studio[19]程序包中的Discover模块, 在COMPASS[20]力场下进行优化, 使能量极小化, 消除内应力。之所以选择COMPASS力场进行MD模拟, 是因为该力场能在较大范围内对处于孤立体系和凝聚态体系的多种物质的构型、振动光谱和热力学等性质同时进行准确预报[21-23], 特别是因为HMX和其它硝胺类化合物包括以硝胺炸药为基的PBXs, 已在该力场中进行过成功的模拟计算, 证明该力场对该类物质很适用[10, 24, 25]。
优化后各结构在NPT系综和温度295 K下进行MD模拟。初始原子运动的速度是按照Maxwell-Boltzman分布确定。牛顿运动方程的求解建立在周期性边界条件、时间平均等效于系综平均等基本假设之上, 积分采用Verlet方法。模拟过程中温度和压力的控制分别采用Anderson[26]和Parrinello方法[27], 范德华(vdW)和静电作用(Coulomb)分别用atom-based和Ewald[28]加和方法, 截断半径取0.95 nm, 并进行截断尾部校正。对于模型Ⅰ、Ⅱ和Ⅲ进行400 ps的MD模拟, 前200 ps用于获得体系的平衡, 后200 ps用于统计分析, 每10 fs取样一次, 共得到20000帧轨迹。而对于模型Ⅰ′、Ⅱ′和Ⅲ′进行1 ns的MD模拟, 前0.5 ns用于获得体系的平衡, 后0.5 ns用于统计分析, 每10 fs取样一次, 共得到50000帧轨迹。
3 结果与讨论 3.1 平衡判别和平衡结构体系平衡必须同时达到温度和能量的平衡。以295 K时NPT系综下β -HMX(4×4×4)超晶胞(模型Ⅰ′)的MD模拟为例, 图 2示出其温度平衡曲线。由图 2可见, 温度上下波动约为±15 K左右, 表明确已达到温度平衡。图 3给出能量随模拟时间波动曲线, 由图 3可见, 体系能量波动亦渐趋平缓, 能量偏差也较小, 表明该体系已达能量平衡。

|
图 2 温度随模拟时间变化的曲线 Fig.2 Curves of temperature vs. simulation time |

|
图 3 能量随模拟时间波动曲线 Fig.3 Curves of energy vs. simulation time |
表 1列出MD模拟所得模型Ⅰ和Ⅰ′平衡结构下的晶胞参数及其实验值。由表 1可见, MD模拟所得结果与实验值非常接近, 其中有些参数(a, α, γ)与实验值几乎相等, 而b、c、β、ρ和V与实验值的相对误差分别仅为3.5%、5.2%、0.4%、2.1%和2.5%。再次表明MD模拟选用的COMPASS力场是合适的, 也说明模拟所得平衡结构是可信的。以模型Ⅰ的平衡结构为例, 如图 4所示。
| 表 1 模型Ⅰ和Ⅰ′的MD晶胞参数及其实验值 Tab.1 Lattice parameters simulated by MD compared with experimental values of β -HMX |

|
图 4 模型Ⅰ的平衡结构 Fig.4 Equilibrium configuration of model Ⅰ |
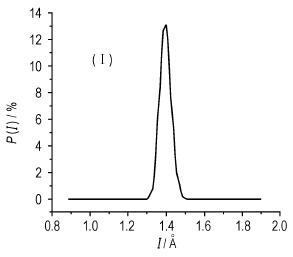
根据MD模拟平衡结构和原子运动轨迹, 原则上可以求得体系的各种性能。图 5仅给出模型Ⅰ、Ⅱ和Ⅲ的N—N引发键的键长分布。表 2给出各模型下N—N键的最可几键长(Lprob)、平均键长(Lave)和最大键长(Lmax)。表 3示出各模型引发键中两个N原子间的相互作用能(EN—N)。基于MD模拟轨迹由波动法分析所得各模型的力学性能列于表 4中。

|
图 5 HMX晶体模型Ⅰ、Ⅱ和Ⅲ平衡体系中的N—N键长分布 Fig.5 N—N trigger bond length distribution in model Ⅰ, Ⅱand Ⅲ of HMX crystals |
| 表 2 HMX各模型中N—N键的Lprob、Lave和Lmax Tab.2 Lprob、Laveand Lmax of trigger N—N bond in STX models of HMX crystals |
| 表 3 各模型中N—N相互作用能及其相关能量 Tab.3 Energy EN—N and correlation energy in six models for HMX crystals |
| 表 4 HMX各模型下的力学性能 Tab.4 Mechanical properties in every each of HMX crystals |
通常实验或量子化学计算, 往往只能给出平均键长, 而MD模拟能给出统计的键长分布, 这是MD模拟的优势所在。比较图 5和表 2可见, 模型Ⅰ、Ⅱ和Ⅲ的引发键键长分布均呈近似对称的高斯分布, 最可几键长与平均键长近似相等。模型Ⅰ、Ⅱ、Ⅲ和Ⅰ′、Ⅱ′和Ⅲ′的平均键长(Lave)均为0.1398 nm, 与HMX的实验键长[17](0.1364 nm)比较接近, 绝对误差和相对误差分别只有0.0034 nm和2.5%。N—N键最大键长变化稍大:比较不切割模型(模型Ⅰ和Ⅰ′)、切割深度1(模型Ⅱ和Ⅱ′)和切割深度1.5(模型Ⅲ和Ⅲ′), 发现体系中原子数多则Lmax稍大些; 不同超胞下模型Ⅰ、Ⅱ和Ⅲ和模型Ⅰ′、Ⅱ′和Ⅲ′, 是否切割分面似乎变化不大。各模型下的N—N引发键最大键长, 其数值虽不尽相同, 也未见有明显递变规律, 但在6个数值中, 最大值(0.1606 nm)与最小值(0.1563 nm)之间仅相差0.0043 nm。文献中已运用Lmax去关联热和撞击感度, 因为具有Lmax的分子虽很少, 但最为活泼, 易于引发分解和起爆[15, 29-31]。这里的研究表明, 取不同模型进行MD模拟所得Lmax变化很小, 不会影响对感度相对大小的判别。当然, 模型Ⅰ′、Ⅱ′和Ⅲ′的引发键键长分布, 也呈近似对称的高斯分布, 为节省篇幅, 本文没有给出。
3.3 引发键连双原子作用能EN—N定义HMX晶体引发键N—N中两个N原子之间的相互作用能为:
| $ {E_{{\rm{N-N}}}} = \left( {E\_E'} \right)/n $ |
式中, ETotal为体系在所用力场框架下的总能量, J·mol-1; E′是固定所有N原子而求得的体系能量, J·mol-1; n是体系中N—N引发键的数目,可见EN—N代表N与N之间的相互作用能, J·mol-1。从表 3可以明显看出,随着体系中原子数增多,其中, N—N相互作用能EN—N有规律地呈单调增大趋势。切割或不切割超晶胞导致的对引发键连双原子作用能的大小无明显影响。EN—N主要由E′(即固定所有N原子而求得的体系能量)特别是由静电能分量所造成。因固定N原子后,静电能的计算随体系中原子数增大而单调增大了。EN—N可用于度量引发键断裂的难易,与热和撞击感度相关联[30-31]。这启示我们,后续MD模拟以引发键连双原子作用能表征感度相对大小时,所取得比较体系的大小(原子数)要相近或相等才有意义。
3.4 弹性力学性能力学性能是含能材料最重要的性质之一。剪切模量越大,表明硬度、屈服强度越大;体模量越大,表明断裂强度越大;C12-C44为柯西压,柯西压若为负值则材料显脆性,若是正值则表明材料延展性较好,其值越高表明体系的延展性越好。表 4给出MD模拟弹性系数、工程模量和泊松比。比较这些弹性力学性能参数,发现各模型泊松比相等,弹性模量变化较小,但切割分面模型(模型Ⅱ、Ⅲ、Ⅱ′和Ⅲ′)的柯西压与原来相应切割前模型相比由负值变为正值。
4 结论综合上述β -HMX不同超晶胞大小和形状、以及是否切割分面和切割深度不同所得各种模型下的MD模拟结果,可以获得如下结论:
(1) β -HMX不同超胞经MD模拟所得各晶胞参数(a、b、c、α、β和γ以及密度ρ和体积V)与实验值非常接近。说明力场选择合适,本模拟所得平衡结构可信。
(2) MD模拟能提供键长统计分布。所得HMX晶体中引发键N—NO2的平均键长与实验值一致,且不随模型改变而变化。
(3) 随体系中原子数目增加,HMX晶体中引发键连双原子作用能EN—N单调增加,EN—N主要由E′(即固定所有N原子而求得的体系能量)特别是由静电能分量所造成。
(4) 除柯西压外,由各模型获得的力学性能数据相接近。
| [1] |
Bedrov D, Simth G D, Sewell T D. Molecular dynamics simulations of HMX crystal polymorphs using a flexible molecule force field[J].
Journal of Comuter-Aided Materials Design, 2001, 8: 77-85. DOI:10.1023/A:1020046817543 |
| [2] |
Sewell T D, Menikoff R, Bedrov D, et al. A molecular dynamics simulation study of elastic properties of HMX[J].
Chem Phys, 2003, 119: 7417 |
| [3] |
肖继军, 黄辉, 李金山, 等. HMX热膨胀系数的分子动力学模拟研究[J].
含能材料, 2007, 15(6): 622-625. XIAO Ji-jun, HUANG H, LI Jin-shan, et al. A MD simulation study of the coefficients of thermal expansion for β -HMX crystal[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2007, 15(6): 622-625. |
| [4] |
Cui H L, Ji G F, Chen X R, et al. Phase transitions and mechanical properties of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine in different crystal phases by molecular dynamics simulation[J].
Journal of Chemical and Engineering Date, 2010, 55: 3121-3129. DOI:10.1021/je100009m |
| [5] |
Zhou T T, Huang F L. Effects of defects on thermal decomposition of HMX via ReaxFF molecular dynamics simulations[J].
The Journal of Physics Chemical B, 2011, 115(2): 278-287. DOI:10.1021/jp105805w |
| [6] |
Ge N N, Wei Y K, Ji G F, et al. Initial decomposition of the condensed β -HMX under shock waves:molecular dynamics simulations[J].
J Phys Chem B, 2012, 116: 13696-13704. DOI:10.1021/jp309120t |
| [7] |
Xiao J J, FangG Y, Ji G F, et al. Simulation investigations in the binding energy and mechanical properties of HMX-based polymer-bonded explosives[J].
Chin Sci Bull, 2005, 50(1): 21-26. DOI:10.1360/982004-147 |
| [8] |
马秀芳, 赵峰, 肖继军, 等. HMX基多组分PBX结构和性能的模拟研究[J].
爆炸与冲击, 2007, 27(2): 109-115. MA Xiu-fang, ZHAO Feng, XIAO Ji-jun, et al. Simulation on structure and property of HMX-based multi-components PBX[J]. Explosion and Shock Wave, 2007, 27(2): 109-115. DOI:10.11883/1001-1455(2007)02-0109-07 |
| [9] |
Xiao J J, Huang H, Xiao H M, et al. MD simulation study on the mechanical properties of HMX crystals and HMX/F2311 PBXs[J].
Acta Chim Sinica, 2007, 65: 1746-1750. |
| [10] |
马秀芳, 肖继军, 李金山. HMX和HMX/HTPB PBX的晶体缺陷理论研究[J].
化学学报, 2008, 66(80): 897-901. MA Xiu-fang, XIAO Ji-jun, LI Jing-shan. A theoretical study on crystal defect of HMX and HMX/HTPB PBX[J]. Acta Chim Sinica, 2008, 66(80): 897-901. |
| [11] |
Xiao J J, Huang H, Xiao H M, et al. Computation of interface interactions and mechanical properties of HMX-based PBX with Estane 5703 from atomic simulation[J].
Mater Sci, 2008, 43: 5685-5691. DOI:10.1007/s10853-008-2704-0 |
| [12] |
Zhu W, Wang X J, Xiao J J, et al. Molecular dynamics simulations of AP/HMX composite with a modified force field[J].
Hazard Mater, 2009, 167: 810-816. DOI:10.1016/j.jhazmat.2009.01.052 |
| [13] |
Xiao J J, Wang W W, iao H M. Study on the relations of sensitivity with energy properties for HMX and HMX-based PBXs by molecular dynamics simulation[J].
Physica B, 2012, 407: 3504-3509. DOI:10.1016/j.physb.2012.05.010 |
| [14] |
Xiao J J, Wang WR, Xiao H M. Study on structure, sensitivity and mechanical properties of HMX and HMX-based PBXs with molecular dynamics simulation[J].
Computational and Theoretical Chemistry, 2012, 999: 21-27. DOI:10.1016/j.comptc.2012.08.006 |
| [15] |
肖继军, 马秀芳, 黄玉成, 等. TATB/氟聚物PBX力学性能的分子动力学模拟[J].
含能材料, 2004, 12: 488-492. XIAO Ji-jun, MA Xiu-fang, HUANG Yu-chen, et al. Molecular dynamics simulation of mechanical properties of TATB/fluorine-polymers PBX[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12: 488-492. |
| [16] |
Xiao J J, Huang Y C, Hu Y J, et al. Molecular dynamics simulation of mechanical properties of TATB/fluorine-polymer PBXs along different surfaces[J].
Sci China B Chem, 2005, 48(6): 504-510. DOI:10.1360/042004-61 |
| [17] |
Chang S C, Henry P B. A study of the crystal structure of β -cyclotetramethylene tetranitramine by neutron difraction[J].
Acta Cryst B, 1970, 26: 1235-1240. DOI:10.1107/S0567740870003941 |
| [18] |
Lewis L, Stevens, Craig J, et al. The elastic constants and related properties of β -HMX determined by Brillouin scattering[J].
Chemical Physics, 2005, 122: 174701 |
| [19] |
Discover, Accelry, San Diego, CA. Materials Studio 4. 2. [CP]. 2008.
|
| [20] |
Sun H. COMPASS: An ab initio forcefield optimized for condensed-phase application-overview with details on alkane and benzene Compounds[J].
J Phys Chem B, 1998, 102: 7338-7364. DOI:10.1021/jp980939v |
| [21] |
Rigby D, Sun H, Eichinger B E. Computer simulations of poly(ethylene oxide): forcefield, PVT diagram and cyclization behavior[J].
Polym Int, 1997, 44: 311-330. DOI:10.1002/(ISSN)1097-0126 |
| [22] |
Sun H, Ren P, Fried J R. The COMPASS force field: parameterization and validation for polyphosph-azenes[J].
Comput Theor Polym Sci, 1998, 8: 229-246. DOI:10.1016/S1089-3156(98)00042-7 |
| [23] |
Bunte S W, Sun H. Molecular modeling of energetic materials: the parameterization and validation of nitrate esters in the COMPASS forcefield[J].
Phys Chem B, 2000, 104: 2477-2489. DOI:10.1021/jp991786u |
| [24] |
Xu X J, Xiao H M, Xiao J J, et al. Molecular dynamics simulations for pure ε-CL-20 and ε-CL-20-based PBXs[J].
Phys Chem B, 2006, 110: 7203-7207. DOI:10.1021/jp060077v |
| [25] |
Qiu L, Xiao H M, Zhu W H, et al. Ab initio and molecular dynamics studies of crystalline TNAD (trans-1, 4, 5, 8-tetranitro-1, 4, 5, 8-tetraazadecalin)[J].
Phys Chem B, 2006, 110: 10651-10661. DOI:10.1021/jp061707w |
| [26] |
Andersen H C. Molecular dynamics simulations at constant pressure and/or temperature[J].
Chem Phys, 1980, 72(4): 2384-2393. |
| [27] |
Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method[J].
Appl Phys, 1981, 52(12): 7182-7190. DOI:10.1063/1.328693 |
| [28] |
Allen M P, Tildesley D J.
Computer Simulation of Liquids[M]. Oxford: Oxford University Press, 1987 |
| [29] |
朱伟, 肖继军, 郑剑, 等. 高能混合物的感度理论判别——不同配比和不同温度AP/HMX的MD研究[J].
化学学报, 2008, 66(23): 2592-2596. ZHU Wei, XIAO Ji-jun, ZHENG Jian, et al. A theoretical criterion for sensitivity of energetic composites——molecular dynamics studies on AP/HMX systems at various concentrations and temperatures[J]. Acta Chimica Sinica, 2008, 66(23): 2592-2596. DOI:10.3321/j.issn:0567-7351.2008.23.004 |
| [30] |
肖鹤鸣, 朱卫华, 肖继军, 等. 含能材料感度判别理论研究——从分子、晶体到复合材料含能材料[J].
含能材料, 2012, 20(5): 514-527. XIAO He-ming, ZHU Wei-hua, XIAO Ji-jun, et al. Theoretical studies on sensitivity criterion of energetic materials——from molecules, crystals, to composite materials[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(5): 514-527. |
| [31] |
赵丽, 肖继军, 陈军, 等. RDX基PBX的模型、结构、能量及其与感度关系的MD研究[J].
中国科学:化学, 2012, 42(1): 1-9. ZHAO Li, XIAO Ji-jun, Chen Jun, et al. Molecular dynamics study on the relationships of modeling, structural structure and energy properties with sensitivity for RDX-based PBXs[J]. Scientia Sinica, 2012, 42(1): 1-9. |
| [32] |
ZAUG J M. In elastic constants of β -HMX and tantalum, eauations of state of supercritical fluids and fluid mixtures and thermal transport determinations[C]//Proceedings of 11th International Detonation Symposium, Snowmass, CO, 1988: 498-509.
|
Six different β -HMX crystalline models were simulated at 295K by molecular dynamics(MD) using COMPASS force field in the isothermal-isobaric (NPT) ensemble.The trigger bond length, the interaction energy between two atoms of trigger bond and the mechanical properties of the HMX crystal were presented and analyzed.