




呋咱醚类化合物是呋咱类含能化合物中重要的一类, 具有能量密度高、标准生成焓大、熔点低、氢含量少(或者无氢)、增塑性强等特点, 可以作为熔铸炸药或含能增塑剂使用, 目前已成为国内外含能材料研究领域的热点之一[1-9]。3, 3′-二(1-氟代-1, 1-二硝基甲基)二呋咱基醚(FOF-13), 是一种典型呋咱醚类含能材料, 其熔点为48 ℃, 密度为1.97 g·cm-3, 氧化系数ko为100%, 标准生成焓ΔHf为146.5 kJ·mol-1, 分解点>270 ℃, 具有高能量密度, 低撞击感度和良好的热稳定性。此外, FOF-13不仅与其它呋咱类含能材料有着很好的相容性, 而且与火炸药常规组分(梯恩梯、黑索今、奥克托今、硝化棉、高氯酸铵、二硝酰胺铵等)的相容性也较好, 其综合性能优于二(2-氟代-2, 2-二硝基乙基)甲缩醛, 是一种性能优良的含能增塑剂[10]。
1998年, Sheremetev在29th ICT年会报道了FOF-13的合成路线[10], 但由于保密原因, 至今仍未披露具体合成实验条件及结构表征数据。探究FOF-13合成路线, 发现肟化反应条件难以控制, 合成技术难度较大, 是合成FOF-13的关键步骤。
本课题组参考文献[10], 以3-氰基-4-硝基呋咱为原料, 自行设计了合成条件, 经分子间醚化、氰基加成、重氮化、双肟化高收率得到了FOF-13关键中间体3, 3′-二(N-羟基偕胺肟基)二呋咱基醚(DOFOF)。重点突破了肟化合成技术, 掌握肟化反应的关键技术, 为进一步合成FOF-13奠定基础, 同时, 亦为自主设计新型含能化合物提供理论依据。研究还考察了氰基加成、双肟化反应的影响因素, 确定了适宜的反应条件; 首次实验发现DOFOF在碱性条件下易发生歧化反应, 并初步提出了N-羟基偕胺肟基发生歧化反应的机理, 为改进合成条件奠定了基础。
2 实验部分 2.1 实验仪器与试剂ZF-Ⅱ型三用紫外仪, 上海市安亭电子仪器厂; NEXUS870型傅里叶变换红外光谱仪, 美国热电尼高力公司; AV500型(500 MHz)超导核磁共振仪, 瑞士BRUKER公司; Vario EL-Ⅲ型元素分析仪, 德国EXEMENTAR公司; LC-2010A液相色谱仪, 日本岛津公司; X-6型显微熔点测定仪, 北京泰克仪器有限公司。
浓硫酸, 分析纯, 西安市福晨化学试剂有限公司; 异丙醇, 分析纯, 天津化学试剂有限公司; 氢氧化钾, 盐酸羟胺, 均为分析纯, 成都市科龙化工试剂厂; 亚硝酸钠, 分析纯, 天津市百世化工有限公司; 乙腈, 色谱纯, SK Chemcials; 3-氰基-4-硝基呋咱(CNNF)自制[8]。
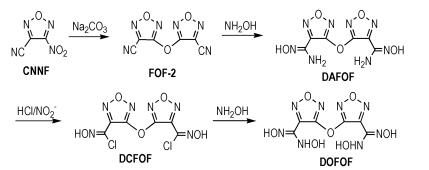
2.2 实验原理合成路线见Scheme 1。

|
Scheme1 Synthetic routes of DOFOF |
室温下将30.00 g (0.214 mol)3-氰基-4-硝基呋咱(CNNF), 18.00 g(0.170 mol)无水碳酸钠加入到1.6 L无水乙腈中, 缓慢升温至78 ℃时, 开始产生黄色硝烟, 回流反应至不再产生黄色硝烟, 减压蒸馏除去乙腈得到淡黄色晶体, 水洗2~3次, 真空干燥, 得白色晶体19.91 g, 收率90.9 %, 纯度99.7 %, m.p.: 68.0~69.0 ℃; 13C NMR (DMSO-d6, 125 MHz), δ: 161.13(C—O), 129.09(C—C=N), 106.63(C≡N); IR (KBr, cm-1), ν: 2270(C≡N), 1563, 1506, 1250, 1032(呋咱环); Anal.calcd for C6N6O3: C 35.31, N 41.18; found C 35.12, N 41.17。
2.3.2 DAFOF的合成冰水浴下, 将4.16 g (20 mmol)FOF-2和2.85 g (41 mmol)NH2OH·HCl加入到50 mL水和25 mL异丙醇的混合溶液中, 搅拌均匀后分批加入2.28 g (21.5 mmol)无水碳酸钠, 升温到25 ℃并保温1 h。过滤除去滤液, 水洗, 真空干燥, 得到白色粉末状固体5.02 g, 收率91.2%, 纯度98.7%, m.p.: 203~204 ℃; 1H NMR (DMSO-d6, 500 MHz), δ: 10.67 (s, 2H, OH), 6.28(s, 4H, NH2); 13C NMR (DMSO-d6, 125 MHz), δ: 160.31(C—O), 142.23(C—C=N), 141.33(C—NH2); IR (KBr, cm-1)ν: 3495, 3454, 3349 (NH2), 3172, 2919 (OH), 1680, 1656, 1525, 1193 (呋咱环); Anal.calcd For C6H6N8O5: C 26.67, N 41.18, H 2.24; found C 26.22, N 41.07, H 2.08。
2.3.3 DCFOF的合成室温下, 将5.40 g(20 mmol)DAFOF加入到30 mL水和55 mL浓盐酸的混合溶液中, 升温搅拌至全溶, 缓慢降温至0 ℃, 滴加2.83 g(41 mmol)亚硝酸钠的饱和溶液, 控制约2 h滴加完毕, 前30 min反应过程剧烈, 冒出大量气泡, 可用少量乙醇作消泡剂; 滴加完毕保温2.5 h, 随后缓慢升温至15 ℃反应2 h。过滤, 水洗, 真空干燥, 得白色粉末状固体5.73 g, 粗品收率92.7 %, 粗品纯度94.6%, 水和乙醇混合重结晶得DCFOF 5.35 g, 收率86.5%, 纯度99.4%。m.p.: 60~61 ℃; 1H NMR(DMSO-d6, 500 MHz), δ: 13.71(s, 2H, OH); 13C NMR(DMSO-d6, 125 MHz), δ: 158.99(C—O), 143.03(C—C=N), 123.40(C—Cl); IR (KBr, cm-1)ν: 3533, 3167, 3020 (OH), 1617, 1570, 1518, 1024 (呋咱环); Anal.calcd For C6H2N6O5Cl2: C 23.32, N 27.20, H 0.65; found C 23.30, N 26.85, H 0.73。
2.3.4 DOFOF的合成20 ℃下, 将1.39 g(20 mmol)盐酸羟胺和1.12 g (20 mmol)氢氧化钾加入到50 mL甲醇中, 过滤除去氯化钾沉淀得羟胺的甲醇溶液, 向其中加入1.55 g(5 mol)DCFOF, 反应2 h后, 减压蒸馏除去溶剂, 将所得粘稠状液体溶于水中, 25 mL乙醚萃取3次, 合并有机相并用无水硫酸镁干燥, 蒸除溶剂后得灰白色粘稠状液体, 静置一段时间后凝结成白色块状固体1.27 g, 粗品收率89.7%, 粗品纯度84.6%。经柱色谱提纯后得DOFOF 1.01 g, 收率71.2%, 纯度98.9%。m.p.: 109~110 ℃; 1H NMR (DMSO-d6, 500 MHz), δ: 11.18 (s, 2H, NH), 9.04 (s, 2H, OH), 8.89 (s, 2H, OH); 13C NMR (DMSO-d6, 125 MHz), δ:161.12(C—O), 145.32(C—C=N), 141.77(C—NHOH); IR (KBr, cm-1)ν: 3533, 3167, 3020 (OH), 1617, 1570, 1518, 1024 (呋咱环); Anal.calcd For C6H6N8O7: C 23.85, N 37.08, H 2.00; found C 24.50, N 36.48, H 2.13。
2.3.5 DOFOF的歧化反应冰水浴下, 将3.09 g(10 mmol)DOFOF加入到30 mL甲醇中, 缓慢滴加0.56 g氢氧化钾的10 mL甲醇溶液, 待滴加完成后反应2 h, 除去甲醇加入20 mL水中, 乙醚萃取15×3 mL, 合并乙醚萃取液, 无水硫酸镁干燥后减压蒸馏得到0.56 g DAFOF, 纯度98.8%, 表征数据同2.3.2。
3 结果与讨论 3.1 DOFOF歧化反应机理DOFOF结构中肟基具有一定的酸性, 在氢氧化钾存在下, 肟基首先与氢氧化钾进行中和反应得到氧负离子过渡态Ⅰ, 然后通过电荷转移, 失去OH-得到3, 3′-二(偕亚胺亚硝基)二呋咱基醚Ⅱ [11], Ⅰ和Ⅱ通过偶合作用转化成Ⅲ; Ⅲ异构化为Ⅳ, Ⅳ的氮氮单键易于断裂, 水解成歧化产物DAFOF和Ⅴ, Ⅴ会以钾盐的形式存在[12]。实验中很容易分离得到DAFOF, 但未能成功分离得到钾盐产物。推测可能的歧化反应机理见Scheme 2。

|
Scheme2 |
根据文献报道[13-16], 以醇和水做混合溶剂, 氰基与羟胺的加成反应能够顺利进行, 收率高, 纯度好。故本实验将醇与水体积比定为1:2, 反应时间为2.0 h, 反应温度为25 ℃时, 考察了不同的醇与水做混合溶剂对DAFOF收率的影响, 结果见表 1。
| 表 1 溶剂对DAFOF收率的影响 Tab.1 Effect of solvent on yield of DAFOF |
由表 1可以看出, 选用甲醇和水作混合溶剂时,产物纯度最好, 达到99.2%, 正丁醇和水做溶剂则最差, 选用烷烃基长的端羟基醇做溶剂时, 产物纯度和收率都随之降低, 这可能是由于FOF-2在长链醇中的溶解度低, 导致氰基水解, 副反应增加; 当选用异丙醇和水做反应溶剂时, 产物收率最高, 达到91.2%, 纯度介于乙醇和正丁醇之间。综合来看, 选用异丙醇和水做反应溶剂较合适。
3.2.2 异丙醇和水体积比对DAFOF收率的影响反应时间为2.0 h, 反应温度为25 ℃时, 考察异丙醇和水的体积比对DAFOF收率的影响, 结果见表 2。
| 表 2 异丙醇与水体积比对DAFOF收率的影响 Tab.2 Effect of volume ratioe of isopropanol and H2O on yield of DAFOF |
由表 2可知, 随着溶剂中异丙醇量逐渐增多, 产物收率先增加后迅速降低, 在水和异丙醇体积比为21时, 收率最高, 达到91.4%;而产物纯度随异丙醇量增多而提高, 最高达到99.2%。这可能是因为单纯用水做反应溶剂时, 由于FOF-2不溶于水, 游离的羟胺不能和FOF-2很好的接触, 不能使反应进行彻底, 液相色谱也证实了此时产物中残留少许原料, 故纯度最差; 而随着异丙醇用量的增加, FOF-2在反应体系中溶解度增加, 与游离羟胺的加成反应容易进行, 所以产品纯度和收率随之增大; 但当异丙醇和水的体积比达到1:1时, 产物的收率严重下降, 这可能与产物在反应体系中溶解度增大以及促使原料中氰基的水解等有关。因此, 水与异丙醇最佳体积比为2:1。
3.3 DOFOF合成条件探讨 3.3.1 氢氧化钾用量DCFOF与NH2OH·HCl物质的量比为1:4, 反应时间为2 h, 反应温度为25 ℃, 考察了DCFOF与KOH的料比对反应的影响, 实验结果见表 3。
| 表 3 DCFOF与KOH料比对DOFOF收率的影响 Tab.3 Effect of molar ratio of DCFOF and potassium hydroxide on the yield of DOFOF |
由表 3可看出, 产品收率随氢氧化钾的增多而降低, 当n(DCFOF):n(KOH)=1:4时, DOFOF的收率和纯度均较好。究其原因, 氢氧化钾一方面与盐酸羟胺进行中和反应, 生成游离的羟胺, 从而使亲核反应能够顺利进行; 另一方面作为缚酸剂吸收反应过程产生的氯化氢。当n(DCFOF):n(KOH)=1:2时, 氢氧化钾用量不足, 反应不能完全进行, 产物中含有较多原料, 纯度仅为49.6%;当n(DCFOF):n(KOH)为1:8时, 氢氧化钾已过量, 强碱下产物中双肟基团会发生歧化、偶合等副反应, 故碱过量对反应不利, 甚至不能得到产品。当n(DCFOF):n(KOH)=1:4时, 羟胺既能游离出来, 氢氧化钾的量又不多余, 因此保证了反应的顺利进行, 收率和纯度都最好。综合考虑, 最佳DCFOF与KOH物质的量比为1:4。
3.3.2 反应温度DCFOF与NH2OH·HCl、KOH的物质量比为1:4:4, 反应时间2 h, 考察了不同反应温度对DOFOF收率的影响, 实验结果见表 4。
| 表 4 反应温度对DOFOF收率的影响 Tab.4 Effect of reaction temperature on the yield of DOFOF |
由表 4可知, 在0~20 ℃范围内, DOFOF收率和纯度变化不大, 到30 ℃以后收率和纯度都有所下降, 杂质明显增多。因此, 为减少副反应的发生, 最佳反应温度为20 ℃。
4 结论(1) 以3-氰基-4-硝基呋咱为原料, 自行设计合成条件, 经分子间醚化、氰基加成、重氮化、双肟化得到了DOFOF, 总收率达51.0%, 并通过红外、核磁、元素分析等方法对产物结构进行了表征。
(2) 确定了合成DAFOF最佳条件:反应溶剂为异丙醇和水的混合溶剂, 二者体积比为1:2, 25 ℃下反应2 h, 收率91.2%;确定了合成DOFOF最佳条件: n(DCFOF):n(NH2OH·HCl):n(KOH)=1:4:4, 20 ℃下反应2h, 收率89.0%。
(3) 发现了DOFOF在强碱下会发生歧化反应, 并通过光谱学信息鉴定出歧化产物DAFOF, 初步提出了DOFOF发生歧化反应可能的机理。
| [1] |
Olofson R A, Michelman J S. Furazan[J].
Journal of Organic Chemistry, 1965, 30(6): 1854-1859. DOI:10.1021/jo01017a034 |
| [2] |
李战雄, 唐松青, 欧育湘, 等. 呋咱含能衍生物合成研究进展[J].
含能材料, 2002, 10(2): 59-65. LI Zhan-xiong, TANG Song-qing, OU Yu-xiang, et al. Synthesis status of furazano energetic derivatives[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2002, 10(2): 59-65. |
| [3] |
范艳洁, 王伯周, 周彦水, 等. 3, 3'-二氰基-4, 4'-偶氮呋咱(DCAF)合成及晶体结构[J].
含能材料, 2009, 17(4): 385-388. FAN Yan-jie, WANG Bo-zhou, ZHOU Yan-shui, et al. Synthesis and crystal structure of 3, 3' -dicyano-4, 4'-azofurazan (DCAF)[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2009, 17(4): 385-388. |
| [4] |
Pivina T S, Sukhachev D V, Evtushenko A V. Comparative characteristic of energy content calculating methods for the furazan series as an example of energetic materials[J].
Propellants Explosives Pyrotechnics, 1995, 20: 5-10. DOI:10.1002/(ISSN)1521-4087 |
| [5] |
Sheremetev A B, Mantseva E V. Hydroxyfurazans: Outlook to using[C]//32nd International Annual Conference of ICT, 2001, 103/1-4.
|
| [6] |
Sheremetev A B, Kulagina V O, Aleksandrova N S, et al. Dinitro Trifurazans with Oxy, Azo, and azoxy Bridges[J].
Propellants Explosive Pyrotechnics, 1998, 23: 142-149. DOI:10.1002/(ISSN)1521-4087 |
| [7] |
Sheremetev A B, Kharitonova O V, Mel'yana T M, et al. Synthesis of symmetrical difurazanyl ethers[J].
Mendeleev Commun, 1996, 4(6): 141-143. |
| [8] |
范艳洁, 王伯周, 来蔚鹏, 等. 3, 3'-二氰基二呋咱基醚(FOF-2)的合成、表征及量子化学研究[J].
有机化学, 2009, 29(4): 614-620. FAN Yan-jie, WANG Bo-zhou, LAI Wei-peng, et al. Synthesis, characterization and quantum chemistry study on 3, 3'-dicyan-difurazanyl ether (FOF-2)[J]. Chinese Journal of Organic Chemistry, 2009, 29(4): 614-620. |
| [9] |
王锡杰, 廉鹏, 葛忠学, 等. 3, 3'-二硝基双呋咱醚(FOF-1)合成、晶体结构及理论研究[J].
化学学报, 2010, 68(6): 557-563. WANG Xi-jie, LIAN Peng, GE Zhong-xue, et al. Synthesis, crystal structure and theoretical research of 3, 3'-dinitrodifurazanyl ether(FOF-1)[J]. Acta Chim Sinica, 2010, 68(6): 557-563. |
| [10] |
Sheremetev A B. 3, 3-Bis(1-fluoro-1, 1-dinitromethyl)difurazanyl ether[C]//Proc. 29th Int. Annaul Conf. ICT, Karlsruhe, Germany. June 30-July 3, 1998: 58: 1-6.
|
| [11] |
Armand J, Minvielle R M. |
| [12] |
Sheremetev A B, Aleksandrova N S, Suponitsky K Y. Bicentral oxidation of nitrosolic acids:synthesis of 1, 1-dinitroalkanes[J].
Mendeleev Communications, 2010, 20: 215-217. DOI:10.1016/j.mencom.2010.06.011 |
| [13] |
Godovikova T I, Vorontsova S K, Konyushkin L D, et al. 4-Methyl-1, 2, 5-oxadiaole-3-carbonitile in the synthesis of 1, 2, 5-oxadiazolyl-1, 2, 4-oxadiazoles[J].
Russian Chemical Bulletin, 2008, 57(11): 2440-2442. DOI:10.1007/s11172-008-0349-4 |
| [14] |
Godovikova T I, Vorontsova S K, Konyushkin L D. Synthesis of 5-(1, 2, 5-oxadiazol-3-yl)-1H-Tetrazoles from 3-cyano-1, 2, 5-oxadiazoles[J].
Russian Chemical Bulletin, International Edition, 2009, 58(2): 406-409. DOI:10.1007/s11172-010-0023-5 |
| [15] |
Luic M, Florence L, Valerie L, et al. New non-hydroxamic ADAMTS-5 inhibitors based on the 1, 2, 4-triazole-3-thiol scaffold[J].
Bioorganic & Medicinal Chemistry Letters, 2010, 20: 6213-6216. |
| [16] |
Marion F, Matthieu D, Nathalie L G, et al. Ethionamide Boosters. 2. Combining bioisosteric replacement and structure-based drug design to solve pharmacokinetic issues in a series of potent 1, 2, 4-oadiazole ethr inhibitors[J].
J Med Chem, 2012, 55: 68-83. DOI:10.1021/jm200825u |
3, 3′-Bis(N-hydroxy amidoxime)difurazanyl ether was synthesized with a yield of 51.0%, and its disproportionation reaction was also discussed.