




浇注高聚物粘结炸药(PBX)是一种典型的多组分颗粒填充型聚合物复合材料, 其组分中包括含能成分和聚合物粘结剂[1]。含能成分在PBX中占的比率依据配方的不同而各不相同, 一般情况下都在80%~90%范围内以满足PBX应用时的能量需求。聚合物粘结剂组分所占的比例虽很小, 但固化网络的性质及其固化反应动力学决定着PBX的工艺, 力学, 贮存等性能[2]。为了获得较好的性能, 固化反应速率的控制是非常重要的。在实际生产中发现, 当选择不同分子量端羟基聚丁二烯(HTPB)时, 出现不同的固化行为, 而文献报道的关于HTPB/2, 4-甲苯二异氰酸酯(TDI)体系反应动力学的研究大多数侧重于HTPB/TDI配比[3], 催化剂[4]及填料[5]的加入对反应速率, 活化能及反应机理的影响, 有关不同分子量HTPB与异氰酸酯固化反应动力学的研究尚未见报道。
目前有关热固性树脂固化反应动力学的研究方法主要有红外光谱法[6]、化学分析法[7]、动态差示扫描量热(DSC)法[8]等, 其中动态DSC法是测定固化反应动力学广泛使用的方法之一。本实验首先采用粘度法对四种不同分子量的HTPB固化反应初期的粘度变化进行监测, 然后采用动态DSC法, 对两种粘度变化趋势差异较大的HTPB的固化行为, 及固化过程中的动力学参数进行研究, 以期为生产中HTPB型号的选择提供依据。
2 实验部分 2.1 原料HTPB, 80 ℃减压蒸馏4 h后使用, HTPB (M1), 数均分子量为1500 g·mol-1, 羟值为1.5 mmol·g-1; HTPB(M2), 数均分子量为2800 g·mol-1, 羟值为0.78 mmol·g-1; HTPB(M3), 数均分子量为1800 g·mol-1, 羟值为1.44 mmol·g-1; HTPB(M4), 数均分子量为3440 g·mol-1, 羟值为0.61 mmol·g-1, 均产自黎明化工研究院; TDI, 化学纯, 北京化学试剂公司。
2.2 实验方法将M1、M2、M3、M4分别与固化剂TDI按照摩尔比1:1配制, 在带有抽真空的搅拌釜中机械搅拌20 min, 粘度测试采用Brookfield型旋转粘度仪, 转子为SC-31, 测试所得M1、M2、M3、M4体系在60 ℃固化过程中粘度的变化, 当胶液出现拉丝现象, 即认为进入凝胶状态。
树脂基体固化反应过程放热量采用TA DSC 910s型差示扫描量热仪测量, 其基本参数为: N2氛围, 温度范围为室温到270 ℃, 升温速率分别为2.5,5,10,15 ℃·min-1。样品质量为15 mg。
2.3 实验原理利用DSC法对热固性树脂固化反应的分析基于以下基本假设[9]。
(1) 热固性树脂固化反应速率与其放热速率成正比:
| $ {\rm{d}}\alpha /{\rm{d}}t = {\rm{d}}H/\left( {{\rm{d}}t\Delta H} \right) $ | (1) |
式中, α是固化度; t是时间, s; dα/dt是固化速率, s-1; ΔH是固化反应总放热量, J·g-1。
(2) 热固性树脂固化反应动力学的速率方程为:
| $ {\rm{d}}\alpha /{\rm{d}}t = A\exp \left( {-{E_{\rm{a}}}/RT} \right)f\left( \alpha \right) $ | (2) |
式中, A是指前因子, s-1; Ea是固化反应活化能, kJ·mol-1; R是气体常数, 8.314 J·K-1·mol·-1; T是绝对温度, K; f(α)机理函数。
(3) 树脂固化反应的放热量与树脂的固化度成正比。
3 结果与讨论 3.1 不同分子量HTPB在固化过程中的粘度变化HTPB固化体系粘度随时间的变化关系见图 1, 为了清楚地反映HTPB体系测试初始阶段其粘度与时间的关系, 70 min前的粘度变化另外作图得图 2。从图 1可看出分子量比较接近的M1和M3体系, M2和M4体系的粘度变化趋势比较接近。而分子量相距较大的M1和M2体系, M3和M4体系的粘度变化相差较大。从图 1和图 2可看出体系粘度的大小开始主要由HTPB(分子量大的HTPB的初始粘度较大)本身的粘度决定, 随着反应的进行, 粘度的大小由各个体系的反应速率决定, 而分子量不同的HTPB的反应速率是不同的, 所以粘度增长趋势发生显著变化。为了说明分子量不同的HTPB在固化反应过程中反应行为的差异, 选取分子量相差较大的M1体系和M2体系进行分析。

|
图 1 M1, M2, M3, M4体系粘度与反应时间的关系 Fig.1 Relationships between dynamic viscosity and time of M1, M2, M3 and M4 system |

|
图 2 M1, M2, M3, M4体系前70 min的粘度变化 Fig.2 Dynamic viscosity of M1, M2, M3 and M4 system in 70 min |
从图 1可以看出, M1体系的粘度增加迅速, 在178 min时已进入凝胶状态; 而M2体系的粘度增加较慢, 在360 min时慢慢进入凝胶状态。M1和M2体系的粘度方程都符合η =Aekt[10], η为胶液粘度, 当M(分子量)=1500 g·mol-1时, A=0.94678 Pa.s, k=0.03319 s-1; M=2800 g·mol-1时, A=2.09761 Pa·s, k=0.0153 s-1。
3.2 不同分子量HTPB的固化反应动力学 3.2.1 DSC的测试结果HTPB的固化反应在DSC曲线上表现为单一的放热曲线, 见图 3(M1体系)和图 4(M2体系)。

|
图 3 M1体系在不同升温速率下的DSC曲线 Fig.3 DSC curves of M1 system at different heating rates |

|
图 4 M2体系在不同升温速率下的DSC曲线 Fig.4 DSC curves of M2 system at different heating rates |
由图 3和图 4可以看出, 随着升温速率的增大, 放热峰温度向高温方向移动, 且放热峰逐渐变陡, 固化时间缩短且体系的放热量逐渐增加。其原因为:随着升温速率的增大, dH/dt变大, 即单位时间产生的热效应增大, 温度差增加, 从而使热固性树脂固化反应放热峰向高温方向移动[11], 固化反应温度升高, 固化时间缩短。表 1是图 3和图 4中固化峰所对应的起始温度, 峰顶温度, 峰终温度及固化放热量数据。
| 表 1 M1和M2体系在不同升温速率下DSC曲线的特征峰温及固化反应热 Tab.1 Characteristic temperature and curing heats of M1and M2 system from DSC curves at different heating rates |
计算固化反应体系的动力学参数主要有Kissinger法[12], Ozawa法[13], Crane法[14]3种。
Kissinger公式:
| $ {\rm{d}}\left[{\ln \left( {\beta /T_{\rm{p}}^2} \right)} \right]/{\rm{d}}\left( {T_{\rm{p}}^{ -1}} \right) = -{E_{\rm{a}}}/R $ | (3) |
Ozawa公式:
| $ {\rm{d}}\left( {\ln \beta } \right)/{\rm{d}}\left( {T_{\rm{p}}^{-1}} \right) =-1.0516{E_{\rm{a}}}/R $ | (4) |
Crane公式:
| $ {\rm{d}}\left( {\ln \beta } \right)/{\rm{d}}\left( {T_{\rm{p}}^{-1}} \right) =-\left( {{E_{\rm{a}}}/nR + 2{T_{\rm{p}}}} \right) $ | (5) |
| $ {E_{\rm{a}}}/nR > > 2{T_{\rm{p}}}, {\rm{d}}\left( {\ln \beta } \right)/{\rm{d}}\left( {T_{\rm{p}}^{-1}} \right) =-{E_{\rm{a}}}/nR $ |
式中,n为反应级数。
常用热固性树脂固化反应模型方程有[15]:
n级模型
| $ {\rm{d}}\alpha /{\rm{d}}t = k{\left( {1-\alpha } \right)^n} $ | (6) |
自催化模型
| $ {\rm{d}}\alpha /{\rm{d}}t = k{\alpha ^{\rm{m}}}{\left( {1-\alpha } \right)^n} $ | (7) |
式中,m是反应级数; k是反应速率常数, s-1; 服从Arrhenius关系k=Aexp(-Ea/RT)。
假设端羟基聚丁二烯的固化反应动力学为n级反应动力学模型, 则其动力学方程如下:
| $ {\rm{d}}\alpha /{\rm{d}}t = A\exp \left( {-{E_{\rm{a}}}/RT} \right){\left( {1-\alpha } \right)^n} $ | (8) |
以ln(β /TP2)和-ln β分别对1000/TP作图, 通过线性拟合可得到一条直线, 见图 5和图 6。从图可看出,ln(β /TP2)和-ln β对1000/TP的线性相关系数都接近0.99。结合Kissinger公式和Crane公式求得M1体系固化反应的Ea=55.87 kJ·mol-1, n=0.88, A=4.70x104 s-1; M2体系固化反应的Ea=60.77 kJ·mol-1, n=0.89, A=1.07 105 s-1。将上述参数带入方程(8)可得:

|
图 5 M1体系-ln β和ln(β /TP2)分别对1000/TP的拟合关系曲线 Fig.5 Linear fitting of caculated -ln β and ln(β /TP2) with 1000/TP for M1 system |

|
图 6 M2体系-ln β和ln(β /TP2)分别对1000/TP的拟合关系曲线 Fig.6 Linear fitting of caculated -ln β and ln(β /TP2) with 1000/TPfor M2 system |
M1体系的固化反应动力学方程为:
| $ {\rm{d}}\alpha /{\rm{d}}t = 4.70 \times {10^4}\exp \left( {-6719/T} \right){\left( {1-\alpha } \right)^{0.88}}; $ |
M2体系的固化反应动力学方程为:
| $ {\rm{d}}\alpha /{\rm{d}}t = 1.07 \times {10^5}\exp \left( {-7309/T} \right){\left( {1-\alpha } \right)^{0.89}}; $ |
图 7和图 8是升温速率为5 ℃·min-1时通过n级固化反应动力学模型计算的dα/dt-T的关系曲线与实验得到的曲线, 以验证n级固化反应动力学模型的合理性。从图可看到模型计算值与实验值的大体趋势较好吻合, 也与Kim J. H.的异氰酸酯组分和多元醇组分的反应模型研究结论一致, 只是n值有所变化[16]。因此该模型能够有效地描述HTPB的固化反应过程, 通过此模型, 可以较好地预测固化度α与温度T和时间t的关系。

|
图 7 M1体系实验值和模型计算值的比较 Fig.7 dα/dt-T curves of experimental and caculated for M1 system at 5 ℃·min-1 |

|
图 8 M2体系实验值和模型计算值的比较 Fig.8 dα/dt-T curves of experimental and model values for M2 system at 5 ℃·min-1 |
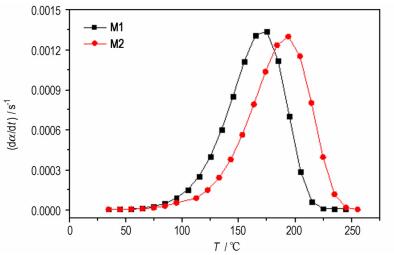
图 9是通过非等温DSC法得到的M1体系和M2体系在升温速率为5 ℃·min-1时, 固化反应速率与温度的关系图, 从图可看出在M1体系固化温度之前, 相同的固化温度时M1体系比M2体系的固化反应速率要大, 当M1体系达到固化特征温度后固化反应速率逐渐减小, 此时M2体系还未达到最大固化速率, 处于继续增加阶段。从峰顶的固化速率可看出M1体系比M2体系的最大固化反应速率要大。结合图 1的粘度测试数值可得出在实际工艺温度时M1体系比M2体系的固化反应速率大, 这主要是因为M1体系的分子量小, 在反应体系中当固化参数为1时, M1体系有更好的活动能力, 能更加自如地接触到固化剂随之发生反应。

|
图 9 M1和M2体系固化速率的比较 Fig.9 Comparison of curing velocity of M1and M2 system |
反应级数决定物质的反应机理, 表观活化能决定固化体系反应活性的大小, 也决定着固化反应能否顺利进行。从前面的计算结果可知M2体系的反应级数比M1体系的略高, 但相差不大, 说明只是分子量的改变并不会改变HTPB固化反应的机理; M2体系的表观活化能(60.77 kJ·mol-1)比M1体系表观活化能(55.87 kJ·mol-1)大, 也即M1体系的反应活性大于M2体系。此数据也进一步说明了M1体系反应速率较大的内在原因。
3.3.3 固化温度及固化反应热从表 1数据可看出, M1体系的特征峰温比M2体系的特征峰温低, 也就是说M1体系比M2体系在更低的温度下就能实现固化。M1体系固化过程的总放热量为134.68 J·g-1(平均值), M2体系固化过程的总放热量为102.40 J·g-1(平均值)。以含10%粘结剂的PBX来进行计算, 假如制备5000 g的PBX, M1体系固化过程中放出的总热量为67.3 kJ, M2体系固化过程中放出的总热量为51.0 kJ。从M1体系和M2体系的放热量可看出, M1体系比M2体系的放热量大, 但放热量差距不大。HTPB体系的放热量比环氧体系的433 J·g-1 [17], 不饱和聚酯树脂体系的282 J·g-1[18]都要小, 这也是HTPB作为PBX粘结剂的优点之一。
3.4 不同分子量HTPB对反应速率影响的机理分析从M1和M2体系的固化反应动力学研究结果可看出, 具有较小分子量的M1体系的固化速率大于分子量较高的M2体系。M1体系具有分子量低的软链段, M2体系具有分子量高的软链段。在硬链段(TDI)含量相同的体系中, 随着软链段分子量的的增加, 软链段的粘度增加, 因此具有大分子量的软链段的扩散速率必然降低, 而软链段的扩散速率的大小直接决定了体系反应速率的大小; 同时具有大分子量的软链段其空间位阻效应相对较大, 位于较大分子量软链段两端的反应基团与位于硬链段上的反应基团的有效碰撞频率相对较低, 从而使反应速率也降低。
4 结论(1) 固化反应开始时, M1体系的粘度小于M2体系, 随着反应的继续进行, 体系的粘度主要由固化反应速率控制, 固化反应速率越大,体系粘度变化越快。M1体系的粘度大于M2体系, 并且M1较早进入凝胶状态。
(2) M1体系的固化反应放热量平均为134.68 J·g-1, M2体系的固化反应放热量为102.40 J·g-1; M1体系固化反应表观活化能为55.87 kJ·mol-1, 反应级数为0.88, 指前因子为4.71×104 s-1, 固化反应动力学方程为dα/dt=4.70×104 exp(-6719/T)(1-α)0.88; M2体系固化反应表观活化能升高为60.77 kJ·mol-1, 反应级数为0.89, 指前因子为1.07×105 s-1, 固化反应动力学方程为dα/dt=1.07×105exp(-7309/T)(1-α)0.89。M1体系固化反应速率大于M2体系固化反应速率。
| [1] |
Valery I L, Bryan F H, Laura B S, et al. Coupled phase transformation, chemical decomposition, and deformation in plastic-bonded explosive: Simulations[J].
Journal of Applied Physics, 2007, 102(10): 1-10. |
| [2] |
Adapaka S K, Vepakomma B R, Rabindra K S, et al. Evaluation of plastic bonded explosive (PBX) formulations based on RDX, Aluminum, and HTPB for underwater applications[J].
Propellants Explosive Pyrotechnics, 2010, 35(4): 359-364. DOI:10.1002/prep.200800048 |
| [3] |
陈清元, 陈中华, 程时远, 等. HTPB/TDI的反应动力学研究[J].
高分子材料科学与工程, 1996, 12(3): 45-49. CHEN Qing-yuan, CHEN Zhong-hua, CHEN Shi-yuan, et al. Study on reaction kinetics of HTPB/TDI by FT-IR[J]. Polymeric Materials Science and Engineering, 1996, 12(3): 45-49. |
| [4] |
金平, 陈建定. HTPB/TDI、HDI聚合反应动力学研究[J].
功能高分子学报, 1998, 11(4): 493-497. JIN Ping, CHEN Jian-ding. Study on reaction kinetics of hydroxylated polybutadiene with tolulene diisocyanates and hexamethylene diisocyanate[J]. Journal of Functional Polymers, 1998, 11(4): 493-497. |
| [5] |
刘晶如, 罗运军. 非等温DSC研究Al/HTPB/TDI体系的固化反应动力学[J].
含能材料, 2009, 17(1): 83-86. LIU Jing-ru, LUO Yun-jun. Curing kinetics of HTPB/TDI/Al System by Non-isothermal DSC[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(1): 83-86. |
| [6] |
Hailu K, Guthausen G, Becker W, et al. In-situ characterization of the cure reaction of HTPB and IPDI by simultaneous NMR and IR measurements[J].
Polymer Testing, 2010, 29(4): 513-519. DOI:10.1016/j.polymertesting.2010.03.001 |
| [7] |
衡明星, 孙培勤, 赵科, 等. 用化学分析法研究三聚氰胺甲醛树脂的结构[J].
粘结, 2007, 28(1): 17-19. HENG Ming-xing, SUN Pei-qin, ZHAO Ke, et al. Study of structure of MF resin by chemical analysis[J]. Adhesion in China, 2007, 28(1): 17-19. |
| [8] |
Korah B C, Kannan K G, Ninan K N. DSC study on the effect of isocyanates and catalysts on the HTPB cure reaction[J].
Journal of Thermal Analysis and Calorimetry, 2004, 78(3): 753-760. DOI:10.1007/s10973-005-0442-0 |
| [9] |
赵卫娟, 张佐光, 孙志杰, 等. 非等温法研究TGDDM /DDS体系固化反应动力学[J].
高分子学报, 2006(4): 564-568. ZHAO Wei-juan, ZHANG Zuo-guang, SUN Zhi-jie, et al. Cure kinetics of TGDDM/DDS system studied by non-isothermal method[J]. Acta polymerica sinica, 2006(4): 564-568. |
| [10] |
郑申声, 关立峰, 董兰, 等. HTPB/N100体系的聚合反应动力学和粘度变化[J].
含能材料, 2011, 19(3): 291-294. ZHENG Shen-sheng, GUAN Li-feng, DONG Lan. Reaction kinetics and viscosity variation of HTPB/N100 polymerization system[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2011, 19(3): 291-294. |
| [11] |
李常雄, 戴亚堂, 胡小平, 等. 非等温DSC法研究TiB2/环氧树脂E-44体系固化动力学[J].
热固性树脂, 2011, 26(3): 6-10. LI Chang-xiong, DAI Ya-tang, HU Xiao-ping, et al. Curing kinetics of TiB2 / epoxy resin E-44 system studied by non-isothermal DSC[J]. Thermosetting Resin, 2011, 26(3): 6-10. |
| [12] |
Kissinge H E. Reaction kinetics in differential thermal analysis[J].
Analytical Chemistry, 1957, 29: 1702-1706. DOI:10.1021/ac60131a045 |
| [13] |
Ozawa T. A new method of analyzing thermo-gravimetric data[J].
Bulletin of the Chemical Society of Japan, 1965, 3(11): 1881-1886. |
| [14] |
Crane L W, Dynes P J, Kaelble D H. Analysis of curing kinetics in polymer composites[J].
Polymer Letter Edition, 1973, 11: 533 DOI:10.1002/pol.1973.130110808 |
| [15] |
刘振海.
热分析导论[M]. 北京: 化学工业出版社, 1991.
LIU Zhen-hai. Journal of thermal analysis[M]. Bei Jing: Chemical Industry Press, 1991 |
| [16] |
Kim J H, Kim S C. Analysis of reaction injection molding process of polyurethane-unsaturated polyester blends[J].
Polymer Engineering and Science, 1987, 27(16): 1243-1251. DOI:10.1002/(ISSN)1548-2634 |
| [17] |
林琳, 宋巍, 李长青. 低粘度环氧树脂体系的固化动力学及其热稳定性[J].
中国表面工程, 2011, 24(5): 73-77. LIN Lin, SONG Wei, LI Chang-qing. Curing kinetics and thermal stability of low viscosity epoxy resin systems[J]. China Surface Engineering, 2011, 24(5): 73-77. |
| [18] |
张宏, 王世兵. 基于DSC的柔性UPR树脂固化反应动力学分析[J].
天中学刊, 2010, 25(5): 9-11. ZHANG Hong, WANG Shi-bing. Researches on curing kinetics of flexible UPR with DSC[J]. Journal of Tian Zhong, 2010, 25(5): 9-11. |
Curing processes of hydroxy-terminated polybutadiene(HTPB)with different molecular weight(M1:1500;M2:2800)/2,4-toluene diisocyanate (TDI) systems were measured by rotational viscometer and non-isothermal differential scanning calorimetry.