




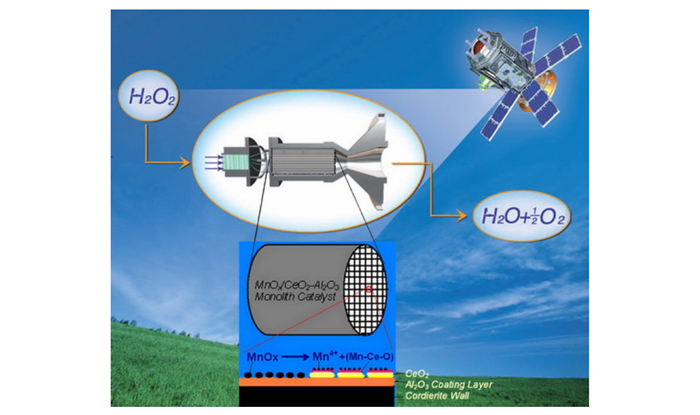
单组元化学推进技术在卫星、飞船和空间站等航天器的轨道和姿态控制及调整中起着至关重要的作用[1]。目前使用的肼单组元推进剂有毒, 能致癌, 严重危害人类健康和安全[2]。随着航天任务的增加, 需要使用更多的单组元推进剂, 为了保障推进剂生产、贮存、操作处理及使用过程中的人员健康和安全, 必须发展无毒单组元推进剂技术[3]。高浓度过氧化氢(HTP)单组元推进剂因其无毒、高密度、可贮存和高能的优势[4], 重新获得了深入研究[5-12, 14-16]。
高浓度过氧化氢在催化剂的作用下分解成高温水蒸气和氧气: H2O2(l)→H2O(g)+1/2O2(g)+热量, 使体积急速膨胀, 把化学能转变成动能。催化剂是HTP单组元推进剂技术的关键。传统颗粒催化剂的缺点是床层流阻大、压降高[13], 在HTP分解过程应用床载(单位截面催化床的推进剂质量流量)低( < 5.5 g·cm-2·s-1)[14], 易磨损和流失[15-16]。蜂窝型整体催化剂床流阻小、压降低[13], 用于HTP分解过程能达到较高的床载, 并且流动分布和传质传热均匀, 耐热冲击和抗磨损性能好[17]。因而, 整体催化剂对高浓度过氧化氢单组元推进剂催化分解具有明显优势。
MnOx是分解过氧化氢的有效活性组分。研究表明, MnO2/Al2O3催化剂分解HTP时很快失活和不稳定[18-19]。添加过渡金属Fe、Zn、Co、Cu和Pb会导致MnO2/Al2O3分解H2O2活性下降或小幅提高[19]。以高锰酸钠为前驱体制备的MnOx整体催化剂在HTP点火后活性组分流失严重[20]。因而, 目前的MnOx负载催化剂, 难以满足HTP单组元推进剂实际应用的需要, 发展高活性和高稳定性的催化剂对高浓度过氧化氢单组元推进剂技术的应用十分必要。
研究表明, CeO2可加速催化剂体相氧的迁移, 提高表面氧的稳定性, 改善过渡金属负载催化剂的储氧和氧化还原性能[21-24], 促进Ni[22, 25]、Cu[26, 29]和Co[27]等与载体间的相互作用和分散, 提高其在甲烷重整[25, 28]、CO氧化[26, 29]和F-T合成[24]等反应的催化活性和选择性。因而, CeO2极好的储存和释放氧的能力及在氧化还原反应中表现的Ce4+/Ce3+离子对的相互转化, 对过渡金属负载催化剂在氧化还原反应中的性能有较大的促进作用。为此本工作研究了CeO2对MnOx/CeO2-Al2O3整体催化剂分解高浓度过氧化氢反应性能的影响。
2 实验部分 2.1 催化剂制备MnOx/CeO2-Al2O3催化剂以乙酸锰和硝酸铈(分析纯)为前驱体, 用浸渍法先在Al2O3载体上负载CeO2, 然后负载MnOx, 经空气下干燥和500 ℃ 3 h焙烧制得。微粒状MnOx/CeO2-Al2O3催化剂中Mn担载量为5%((Mn质量/催化剂质量)×100%), 初始载体为微粒状γ-Al2O3(粒径 < 65 μm), 将MnOx/Al2O3样品标记为MnA, 并按CeO2-Al2O3载体中CeO2负载量((CeO2质量/Al2O3质量)×100%)1%、5%、10%、20%、30%和50%依次将微粒MnOx/CeO2-Al2O3样品标记为Mn1CA、Mn5CA、Mn10CA、Mn20CA、Mn30CA和Mn50CA。整体催化剂的载体是负载了γ-Al2O3涂层(图 1c和1d)(94.9 m2/g)的1075孔/inch2堇青石蜂窝陶瓷(图 1a); MnOx/Al2O3整体催化剂中MnOx含量为7.0%; MnOx/CeO2-Al2O3整体催化剂(图 1b)的MnOx含量为7.0%, CeO2含量为3.1%;整体催化剂直径为14.5 mm, 长度9.5 mm, 发动机热试用的整体催化剂床由20块整体催化剂组合而成。纯MnOx由乙酸锰在400~900 ℃空气下焙烧3 h获得, 标记为MnOx-400 ℃、MnOx-600 ℃和MnOx-900 ℃。

|
图 1 1075孔/inch2蜂窝陶瓷载体、整体催化剂和Al2O3涂层图片 Fig.1 Photographs of 1075 cell/inch2 honeycomb ceramic support, monolith catalyst and Al2O3 coating layer |
催化剂的X射线衍射(XRD)分析在荷兰帕纳科公司(PANalytical)X′ Pert Pro型X射线衍射仪上进行, 以Cu Kα作辐射源, 扫描范围(2θ): 5°~80°, 操作电压40 kV。H2-TPR在美国麦克仪器公司(Micromeritics)AutoChem Ⅱ 2920型全自动化学吸附仪上测定, 还原气为10% H2-Ar混合气, 升温速率10 ℃·min-1, 热导池检测器检测耗H2量。扫描电子显微镜(SEM)图片在日本JEOL公司JSM6360型扫描电镜上获得。
催化剂分解过氧化氢反应的活性用恒压排气量气法在间歇反应器中进行[30], 反应温度30 ℃, 微粒催化剂装量10 mg, 0.47% H2O2溶液体积50 ml, 释放的氧气体积换算为标准状态(0 ℃, 101.3 kPa)。
整体催化剂对高浓度过氧化氢分解反应的稳定性用HTP单组元发动机热试试验来考察[31]。HTP发动机推力为25 N, 推进剂为90% H2O2(杂质含量≤0.02%)。HTP热试装置流程和发动机结构如图 2所示。HTP贮存于内衬聚四氟乙烯的不锈钢贮罐中, 试验时由高压氮气提供推动力, 高压氮气瓶中的氮气经稳压阀进入过氧化氢贮罐, 推动HTP至电磁阀入口, 电磁阀打开, HTP溶液即进入发动机内部的整体催化剂床, 并被催化分解, 分解反应产生的高温水蒸气和氧气经发动机喉管排入大气。HTP点火过程中喷前压(ps, 发动机喷注板前部压力)、室压(pc)(发动机反应室中催化剂床出口处压力)和液体HTP推进剂质量流速(MH2O2)由计算机自动实时记录。

|
图 2 HTP热试系统和发动机示意图 1—氮气瓶, 2, 4, 8, 9—截止阀, 3—稳压阀, 5—泄气口, 6—HTP贮罐, 7—HTP加注排放口, 10—电磁阀, 11—发动机 Fig.2 Schemes of HTP hot test system and thruster 1—N2 gas cylinder, 2, 4, 8, 9—cut-off valve, 3—pressure regulator, 5—gas outlet, 6—HTP container, 7—HTP inlet and outlet, 10—electromagnetic valve, 11—thruster |
图 3为MnOx/CeO2-Al2O3催化剂、γ-Al2O3和CeO2的XRD谱图。当CeO2-Al2O3载体中CeO2负载量((CeO2质量/Al2O3质量)×100%)小于5%, 即MnOx/CeO2-Al2O3催化剂中Mn/(Mn+Ce)摩尔比大于0.78时, MnA和Mn1CA样品XRD图谱中未发现强或尖锐的MnOx衍射信号, 只在42.8°处出现结晶较差的低强度MnO2宽化衍射峰, 并显示了γ-Al2O3特征峰(33.1°、37.5°、39.6°、45.7°、67.3°)。当载体中5%≤CeO2负载量≤20%, 即催化剂中0.78≥Mn/(Mn+Ce)摩尔比≥0.5时, Mn5CA、Mn10CA和Mn20CA样品XRD谱线由逐渐增强的立方萤石结构CeO2衍射峰(28.6°、33.3°、47.6°、56.5°)及逐渐弱化的γ-Al2O3特征峰和MnO2宽化衍射峰(42.8°)组成。而当载体中CeO2负载量超过20%, 即催化剂中Mn/(Mn+Ce)摩尔比低于0.5时, Mn30CA和Mn50CA样品XRD图谱主要由CeO2物相构成, γ-Al2O3和MnO2宽化衍射峰信号很弱。XRD结果表明, MnOx/CeO2-Al2O3的表面结构明显受载体中CeO2负载量的影响, CeO2的添加促进了MnOx在催化剂表面的高分散和形成Mn-Ce-O固溶体。这与Machida等得出的Mn-Ce氧化物晶相组成依赖于Mn/(Mn+Ce)摩尔比的观点相吻合[32]。Mn2O3和CeO2之间在焙烧过程形成了固溶体。由于结构相似, Mn3+在CeO2立方萤石结构中取代Ce4+是可能的[32-33]。与纯CeO2相比, 图 3中MnOx/CeO2-Al2O3的XRD谱图中CeO2特征峰2θ角轻微向高值迁移证实了此结论。这是由于, 焙烧过程使浸渍在CeO2表面的Mn3+(离子半径0.066 nm)扩散至CeO2立方晶格内部, 部分取代半径较大的Ce4+(离子半径0.094 nm), 形成Mn-Ce-O固溶体, 引起单元晶格收缩, 使CeO2立方萤石结构点阵晶面间距和晶胞参数变小(表 1), 从而导致XRD特征峰轻度高移和宽化。CeO2衍射峰在Mn5CA中开始明显出现, 是由于CeO2在Al2O3表面接近单层覆盖(CeO2负载量约为6%)[34], 形成微粒所致; 并且, 随着MnOx/CeO2-Al2O3催化剂中CeO2量的增加, CeO2对Al2O3表面覆盖度、结晶度和晶粒粒径增大, 使CeO2特征信号增强; 而CeO2覆盖度的增大会增强CeO2-MnOx间的相互作用和固溶体形成, 使Mn物种(Mn4+和Mn3+)晶化下降和分散度提高, 导致MnO2衍射峰(42.8°)强度减弱。纯MnOx的XRD结果(图 4)表明, 400 ℃焙烧的MnOx由α-MnO2(18.1°、28.9°、36.1°、59.9°)和较少量α-Mn2O3组成。600 ℃和900 ℃焙烧的MnOx为α-Mn2O3晶相(23.2°、33.0°、38.3°、45.2°、49.4°、55.2°、64.2°、65.8°)。

|
图 3 MnOx/CeO2-Al2O3、γ-Al2O3和CeO2的XRD谱图 Fig.3 XRD patterns of MnOx/CeO2-Al2O3, γ-Al2O3 and CeO2 |
| 表 1 MnOx/CeO2-Al2O3中CeO2晶胞参数 Tab.1 Lattice parameters of CeO2 in MnOx/CeO2-Al2O3 |

|
图 4 纯MnOx的XRD谱图 Fig.4 XRD patterns of pure MnOx |
图 5为MnOx/CeO2-Al2O3催化剂的H2-TPR谱图。由图 5可见, MnOx/Al2O3催化剂(MnA)出现两个相互重叠的H2吸收峰, α峰峰温为287 ℃, β峰峰温为313 ℃, β峰面积略高于α峰。α峰对应Mn物种的低温还原过程: MnO2/Mn2O3→Mn3O4, β峰对应Mn物种的高温还原过程: Mn3O4→MnO[35]。因而, MnOx/Al2O3活性相为Mn2O3和MnO2混合物, MnO2摩尔量稍低于Mn2O3。这与MnOx/Al2O3和纯MnOx的XRD结果相吻合。随着载体中CeO2的添加及其负载量的提高, MnOx/CeO2-Al2O3催化剂的α峰面积增大, β峰面积减小, 表明Mn4+物种量逐渐增大, Mn3+物种量下降。这归因于CeO2表面氧的作用[36]。CeO2的添加增加了载体表面氧的量和移动性, 增强了MnOx与CeO2-Al2O3载体间的相互作用, 使低价态Mn3+被氧化到活泼的高价态Mn4+。XRD结果显示的Mn-Ce-O固溶体的存在, 佐证了MnOx与CeO2-Al2O3间较强的相互作用。随着载体中CeO2负载量提高, MnOx/CeO2-Al2O3催化剂的α峰位置向低温迁移(287 ℃ (MnA)→286 ℃ (Mn1CA)→261 ℃ (Mn5CA)→260 ℃ (Mn10CA)→234 ℃ (Mn20CA)→213 ℃ (Mn30CA)→220 ℃ (Mn50CA))。这是由于, 随着CeO2负载量的提高, 载体表面氧的移动性增大, 使MnOx/CeO2-Al2O3催化剂低温活性中心数和还原能力增大, 促进了Mn4+物种的还原(MnO2→Mn3O4)和催化活性的提高。Mn4+物种以不同的形式分布在MnOx/CeO2-Al2O3催化剂中, 其中包括高分散在Al2O3和CeO2表面的MnO2粒子、嵌进CeO2晶格缺陷位的孤立Mn4+和Mn-Ce-O固溶体中的Mn4+-O-Ce4+[37]。负载量的增加使CeO2对Al2O3表面覆盖度逐渐提高, 从而使分布在Al2O3表面的Mn物种逐渐减少, 而分布在CeO2表面的高分散MnO2粒子增加, 并导致孤立Mn4+和Mn4+-O-Ce4+量显著增加。载体中CeO2负载量低于20%时, MnOx/CeO2-Al2O3催化剂β峰位置变化较小, 表明其主要对应Mn3O4→MnO的还原; 当载体中CeO2负载量超过20%时, β峰面积增至与α峰相当, 并且位置向高温迁移(319 ℃ (Mn10CA)→367 ℃ (Mn50CA))。这归因于Mn3O4→MnO还原峰与CeO2表面氧的还原峰重叠[36]。CeO2表面氧在300~500 ℃被还原, 峰温在480 ℃[38]。高CeO2担载量的MnOx/CeO2-Al2O3催化剂在550 ℃以上出现的还原峰, 为体相CeO2的还原[35-36]。

|
图 5 MnOx/CeO2-Al2O3催化剂的H2-TPR谱图 Fig.5 H2-TPR profiles of MnOx/CeO2-Al2O3 catalysts a—MnA, b—Mn1CA, c—Mn5CA, d—Mn10CA, e—Mn20CA, f—Mn30CA, g—Mn50CA |
图 6是MnOx/CeO2-Al2O3催化剂分解H2O2时释放的氧气体积随时间的变化曲线。如图 6所示, MnA(MnOx/Al2O3)分解H2O2的氧气释放量随时间增加最慢, 表明其催化活性最低。当CeO2-Al2O3载体中CeO2负载量增至5%时, Mn5CA样品分解H2O2速率显著加快。载体中CeO2负载量大于5%后, 催化剂分解H2O2速率增幅变缓。Mn30CA样品分解H2O2反应速率最大, 活性最高。催化剂对H2O2分解反应活性顺序为: MnA < Mn1CA < Mn5CA≈Mn10CA < Mn20CA < Mn50CA < Mn30CA。

|
图 6 MnOx/CeO2-Al2O3上H2O2分解时释放O2体积随时间的变化 Fig.6 O2 volume vs time during H2O2 decomposition on MnOx/CeO2-Al2O3 |
MnOx/CeO2-Al2O3催化剂分解H2O2反应的高活性归因于活泼的低温活性中心Mn4+的增加。XRD和H2-TPR结果表明, CeO2的添加增强了催化剂表面氧的活化和移动性, 使低价态Mn3+易于被氧化到活泼的高价态Mn4+, 提高了低温活性中心数和还原能力, 因而使MnOx/CeO2-Al2O3催化剂活性提高。Mn5CA催化剂表面低温活性中心Mn4+量最大, 与H2O2分解活性显著增加相吻合。载体中CeO2负载量进一步增加后, 催化剂中Mn4+量虽然不再明显增加, 但Mn10CA至Mn50CA催化剂的H2-TPR谱图α峰峰温向更低温度迁移, 显示低温活性中心更活泼, 催化剂活性更高, 因而, 催化剂分解H2O2的活性仍有所增加。其中, Mn30CA的α峰峰温最低, 活性最高。
3.4 整体催化剂的发动机热试性能为了考察催化剂分解高浓度过氧化氢反应的稳定性, 用装填1075孔/inch2整体催化剂的25 N发动机进行了90% H2O2催化分解反应热试。图 7给出了90% H2O2点火过程中室压、喷前压和H2O2质量流速随时间的变化曲线。由图 7a可以看出, MnOx/Al2O3整体催化剂在90% H2O2分解过程的床载达到了18.8 g·cm-2·s-1。室压在点火23 s后开始下降。这是归因于MnOx/Al2O3整体催化剂失活导致的HTP分解效率下降。用MnOx/CeO2-Al2O3整体催化剂在18.5 g·cm-2·s-1床载下进行了两次90% H2O2长程稳态点火, 第一次100 s结果如图 7b所示, 第二次200 s,结果与图 7b相同。两次稳态点火过程中室压和流量稳定, 显示催化活性没有下降, 催化剂工作稳定。

|
图 7 整体催化剂分解90% H2O2时的室压(Pc)、喷前压(Ps)和H2O2质量流速(MH2O2)随点火时间的变化 Fig.7 Variation of chamber pressure (Pc), supply pressure (Ps) and H2O2 mass flow rate (MH2O2) with firing time during 90% H2O2 decomposition on monolith catalysts |
与MnOx/Al2O3整体催化剂相比, MnOx/CeO2-Al2O3整体催化剂分解高浓度过氧化氢反应的催化活性和稳定性显著提高。这是由于, 添加CeO2后, MnOx/CeO2-Al2O3整体催化剂通过表面氧的作用, 将低价态Mn3+氧化到易还原的高价态Mn4+, 使Mn物种低温活性中心数量增加, 并形成丰富的表面活性氧和Ce4+→Ce3+离子对, 从而增强了还原氧化能力, 促进高浓度过氧化氢分解反应, 提高催化剂活性和稳定性。
4 结论CeO2通过形成表面活性氧和Mn-Ce-O固溶体, 改变MnOx/CeO2-Al2O3催化剂表面结构性质和Mn物种价态(Mn3+→Mn4+), 增加了低温活性中心数, 提高了催化剂的还原氧化能力和催化活性。催化剂活性评价和高浓度过氧化氢热试结果表明, CeO2的添加促进了MnOx/CeO2-Al2O3整体催化剂分解高浓度过氧化氢反应活性和稳定性的提高。
| [1] |
Gulczinski F S, Spores R A, Stuhlberger J. In-space propulsion[R]. Air Force Research Laboratory report, AFRL-PR-ED-TP-2003-105: 2003.
|
| [2] |
Wernimont E J. System trade parameter comparison of monopropellants: hydrogen peroxide vs hydrazine and others[R]. AIAA Paper 2006-5236: 2006.
|
| [3] |
Cervone A, Torre L, Agostino L. Development of hydrogen peroxide monopropellant rockets[R]. AIAA Paper 2006-5239: 2006.
|
| [4] |
Ventura M C. Long term storability of hydrogen peroxide. AIAA Paper 2005-4551[R]. 2005.
|
| [5] |
Morlan P, Wu P, Nejad A, et al. Catalyst development for hydrogen peroxide rocket engines. AIAA Paper 1999-2740[R]. 1999.
|
| [6] |
Ventura M, Wernimont E. Advancements in high concentration hydrogen peroxide catalyst beds[R]. AIAA Paper 2001-3250: 2001.
|
| [7] |
Wernimont E, Ventura M. Catalyst bed testing for development of a 98% hydrogen peroxide procurement specification. AIAA Paper 2002-3852[R]. 2002.
|
| [8] |
Beutien T R, Heister S D, Rusek J J, et al. Cordierite-based catalytic beds for 98% hydrogen peroxide. AIAA Paper 2002-3853[R]. 2002.
|
| [9] |
Wernimont E J, Durant D. State of art high performance hydrogen peroxide catalyst beds[R]. AIAA Paper 2004-4147: 2004.
|
| [10] |
Wernimont E J. Monopropellant hydrogen peroxide rocket systems: optimum for small scale[R]. AIAA Paper 2006-5235: 2006.
|
| [11] |
Ventura M, Wernimont E, Dillard J. Hydrogen peroxide -optimal for turbomachinery and power applications[R]. AIAA Paper 2007-5537: 2007.
|
| [12] |
An S, Jo S, Wee J, et al. Preliminary flight test of hydrogen peroxide retro-propulsion module[J].
Acta Astronautica, 2010, 67: 605-612. DOI:10.1016/j.actaastro.2010.04.010 |
| [13] |
Boger T, Heibel A K, Sorensen C M. Monolithic catalysts for the chemical industry[J].
Ind Eng Chem Res, 2004, 43: 4602-4611. DOI:10.1021/ie030730q |
| [14] |
Pasini A, Torre L, Romeo L, et al. High mass flux tests on catalytic beds for H2O2 monopropellant thruster[C]//ESA Space Propulsion Conference, San Sebastian, Spain, 2010.
|
| [15] |
Romeo L, Torre L, Pasini A, et al. Development and testing of Pt/Al2O3 catalysts for hydrogen peroxide decomposition[C]//5th International Spacecraft Propulsion Conference, Heraklion, Greece, 2008.
|
| [16] |
Torre L, Psini A, Romeo L, et al. Firing performance of advanced hydrogen peroxide catalytic beds in a monopropellant thruster prototype[R]. AIAA paper 2008-4937 : 2008.
|
| [17] |
Kappenstein C, Brahmi R, Amariei D, et al. Catalytic decomposition of energetic compounds -influence of catalyst shape and ceramic substrate[R]. AIAA Paper 2006-4546: 2006.
|
| [18] |
Jo S, Jang D, An S, et al. Chugging instability of H2O2 monopropellant thrusters with catalyst reactivity and support sizes[J].
Journal of Propulsion and Power, 2011, 27(4): 920-924. DOI:10.2514/1.B34222 |
| [19] |
田含晶, 张涛, 杨黄河, 等. 用于过氧化氢分解的锰铅复合氧化物催化剂[J].
催化学报, 2000, 21(6): 600-602. TIAN Han-jing, ZHANG Tao, YANG Huang-he, et al. Manganese-lead mixed oxide catalysts for decomposition of hydrogen peroxide[J]. Chinese Journal of Catalysis, 2000, 21(6): 600-602. |
| [20] |
Brahmi R, Batonneau Y, Kappenstein C, et al. Ceramic catalysts for the decomposition of H2O2. Influence of the wash-coat procedure and the active phase[C]//8th International Hydrogen Peroxide Propulsion Conference, Purdue University, 2005: 13-22.
|
| [21] |
卢冠忠, 汪仁. 氧化铈在非贵金属氧化物催化剂中的作用Ⅰ.铜和铈负载型氧化物中的氧的性能[J].
催化学报, 1991, 12(2): 83-90. LU Guan-zhong, WANG Ren. Role of ceria in non-precious metal oxide catalysts Ⅰ. The oxygen properties of Cu and Ce oxides supported on γ-Al2O3[J]. Chinese Journal of Catalysis, 1991, 12(2): 83-90. |
| [22] |
卢冠忠, 汪仁. 氧化铈在非贵金属氧化物催化剂中的作用Ⅱ. Mn-O/γ-Al2O3和Mn-Ce-O/γ-Al2O3催化剂中氧的性能[J].
催化学报, 1991, 12(4): 261-267. LU Guan-zhong, WANG Ren. Role of ceria in non-precious metal oxide catalysts Ⅱ. The oxygen properties of Mn-O and Mn-Ce-O supported on alumina[J]. Chinese Journal of Catalysis, 1991, 12(4): 261-267. |
| [23] |
卢冠忠, 汪仁. 氧化铈在非贵金属氧化物催化剂中的作用Ⅲ.铈对Cu-Mn-O/γ-Al2O3催化剂中氧的性能和固相结构的影响[J].
催化学报, 1991, 12(4): 314-319. LU Guan-zhong, WANG Ren. Role of ceria in non-precious metal oxide catalysts Ⅲ. The oxygen properties and solid structure of Cu-Mn-O and Cu-Mn-Ce-O supported on γ-Al2O3[J]. Chinese Journal of Catalysis, 1991, 12(4): 314-319. |
| [24] |
卢冠忠, 汪仁. 氧化铈在非贵金属氧化物催化剂中的作用Ⅳ.在负载型Cu-Mn-Ce-O催化剂上甲苯的催化燃烧[J].
催化学报, 1992, 13(6): 466-470. LU Guan-zhong, WANG Ren. Role of ceria in non-precious metal oxide catalysts Ⅳ. Catalytic combustion of toluene over Cu-Mn-Ce-O supported on alumina[J]. Chinese Journal of Catalysis, 1992, 13(6): 466-470. |
| [25] |
Wang S, Lu G Q. Role of CeO2 in Ni/CeO2-Al2O3 catalysts for carbon dioxide reforming of methane[J].
Applied Catalysis B: Environmental, 1998, 19: 267-277. DOI:10.1016/S0926-3373(98)00081-2 |
| [26] |
蒋晓原, 周仁贤, 毛建新, 等. CeO2对CuO/Al2O3分散状态及催化性能的影响[J].
分子催化, 1999, 13(3): 176-180. JIANG Xiao-yuan, ZHOU Ren-xian, MAO Jian-xin, et al. Effects of CeO2 on dispersion state and catalytic property of CuO/Al2O3[J]. Chinese Journal of Molecular Catalysis, 1999, 13(3): 176-180. |
| [27] |
代小平, 余长春, 沈师孔. 助剂CeO2对Co/Al2O3催化剂上F-T合成反应性能的影响[J].
催化学报, 2001, 22(2): 104-108. DAI Xiao-ping, YU Chang-chun, SHEN Shi-kong. Promotion effect of ceria on Fischer-Tropsch synthesis performance over Co/Al2O3 catalyst[J]. Chinese Journal of Catalysis, 2001, 22(2): 104-108. |
| [28] |
Cai X, Dong X, Lin W. Effect of CeO2 on the catalytic performance of Ni/Al2O3 for autothermal reforming of methance[J].
Journal of Natural Gas Chemistry, 2008, 17: 98-102. DOI:10.1016/S1003-9953(08)60033-X |
| [29] |
Wang H, Li D, Dai Y, et al. Effect of CO pretreatment on the performance of CuO/CeO2/γ-Al2O3 catalysts in CO+O2 reactions[J].
Applied Catalysis A:General, 2009, 360: 26-32. DOI:10.1016/j.apcata.2009.02.046 |
| [30] |
周志江, 王晓东, 单继宏, 等. 预处理条件对高浓度过氧化氢分解用银网催化剂初始活性的影响[J].
催化学报, 2006, 27(11): 957-960. ZHOU Zhi-jiang, WANG Xiao-dong, SHAN Ji-hong, et al. Effect of pretreatment conditions on initial catalytic activity of silver screen catalyst for the catalytic decomposition of highly concentrated hydrogen peroxide[J]. Chinese Journal of Catalysis, 2006, 27(11): 957-960. DOI:10.3321/j.issn:0253-9837.2006.11.007 |
| [31] |
Tian Han-jing, Zhang Tao, Sun Xiao-ying, et al. Performance and deactivation of Ir/γ-Al2O3 catalyst in the hydrogen peroxide monopropellant thruster[J].
Applied Catalysis A: General, 2001, 210: 55-62. DOI:10.1016/S0926-860X(00)00829-2 |
| [32] |
Machida M, Uto M, Kurogi D, et al. MnOx-CeO2 binary oxides for catalytic NOx sorption at low temperatures. Sorptive removal of NOx[J].
Chemistry of Materials, 2000, 12: 3158-3164. DOI:10.1021/cm000207r |
| [33] |
Wang Z, Shen G, Li J, et al. Catalytic removal of benzene over CeO2-MnOx composite oxides prepared by hydrothermal method[J].
Applied Catalysis B: Environmental, 2013, 138-139: 253-259. DOI:10.1016/j.apcatb.2013.02.030 |
| [34] |
Yao H C, Yao Y F Y. Ceria in automotive exhaust catalyst: Ⅰ. Oxygen storage[J].
Journal of Catalysis, 1984, 86: 254-265. DOI:10.1016/0021-9517(84)90371-3 |
| [35] |
Trawczynski J, Bielak B, Mista W. Oxidation of ethanol over supported manganese catalysts-effect of the carrier[J].
Applied Catalysis B: Environmental, 2005, 55: 277-285. DOI:10.1016/j.apcatb.2004.09.005 |
| [36] |
Tang X, Li Y, Huang X, et al. MnOx-CeO2 mixed oxide catalysts for complete oxidation of formaldehyde: Effect of preparation method and calcination temperature[J].
Applied Catalysis B: Environmental, 2006, 62: 265-273. DOI:10.1016/j.apcatb.2005.08.004 |
| [37] |
Wang X, Kang Q, Li D. Catalytic combustion of chlorobenzene over MnOx-CeO2 mixed oxide catalysts[J].
Applied Catalysis B: Environmental, 2009, 86: 166-175. DOI:10.1016/j.apcatb.2008.08.009 |
| [38] |
Huang Yan-qiang, Wang Ai-qin, Li Lin, et al. "Ir-in-ceria": A highly selectively catalyst for preferential CO oxidation[J].
Journal of Catalysis, 2008, 255: 144-152. DOI:10.1016/j.jcat.2008.01.024 |
The CeO2 addition promotes the activity and stability of MnOx/CeO2-Al2O3 monolith catalyst for catalytic decomposing high concentration hydrogen peroxide monopropellant.