




多硝基金刚烷(PNA)由于含有较高能量密度, 且稳定性较好, 是一类具有较大应用前景的耐热、钝感含能材料, 其能量和爆轰性能随着所含硝基数目的增加而提高[1]。计算表明, 八个硝基金刚烷的输出能量与HMX相当。美国陆军武器研究发展和工程中心(ARDEC)从20世纪80年代开始就注意到多硝基金刚烷, 并将之列入到21世纪发展战略中[2]。多硝基金刚烷的合成多采用间接法合成, 目前合成出的多硝基金刚烷有1, 3, 5, 7-四硝基金刚烷[3]、2, 2-二硝基金刚烷[4]、2, 2, 6, 6-四硝基金刚烷[4]、2, 2, 4, 4-四硝基金刚烷[5]、2, 2, 4, 4, 6, 6-六硝基金刚烷[6]和1, 2, 2-三硝基金刚烷[7]。
金刚烷具有三维笼状碳骨架, 结构紧密, 具有较好的热安定性和起爆感度。2, 2, 4, 4-四硝基金刚烷的密度为1.65 g·cm-3, 熔点为138 ℃[5]。计算表明, 它的生成热为-123.65 kJ·mol-1, 爆速为7350 m·s-1, C-J压力为23.58 GPa[8], 可用于钝感炸药以及推进剂配方中。Dave[5]报道了2, 2, 4, 4-四硝基金刚烷的唯一合成路线, 总收率为4.9%。此合成路线存在以下缺点: (1)关于重要的中间体4, 4-乙撑二氧基金刚烷-2-酮的合成, 内酯重排和氧化反应产率低, 并且内酯重排反应产物和原料分离困难; (2)采用无水硝酸进行酮肟的偕硝化反应, 产率较低。为解决上述问题, 本研究采用新工艺合成了2, 2, 4, 4-四硝基金刚烷, 优化了重要中间体4, 4-乙撑二氧基金刚烷-2-酮的合成方法, 并使用绿色硝化剂五氧化二氮对酮肟进行偕硝化反应, 提高了2, 2, 4, 4-四硝基金刚烷的总收率。
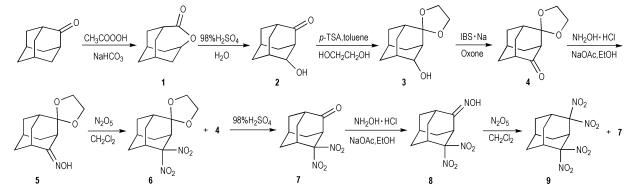
2 实验部分 2.1 合成路线合成路线见Scheme 1。

|
Scheme 1 |
Nicolet Impact 410型傅里叶变换红外光谱仪(美国Thermofisher公司); Bruker Avance-Ⅲ DRX 500 MHz核磁共振仪(德国Bruker公司); Vario EL-Ⅲ型元素分析仪(德国Elemetar公司); WRS-1B数字熔点仪(上海申光仪器仪表有限公司)。
五氧化二氮和邻碘苯磺酸钠分别参考文献[9]和[10]自制, 其它试剂均为市售分析纯, 使用前未经处理。
2.3 合成实验 2.3.1 4-氧杂三环[4.3.1.13, 8]十一烷-5-酮(1)的合成向500 mL三口烧瓶中加入10 g(66 mmol)金刚烷酮、100 mL二氯甲烷和100 mL饱和碳酸氢钠水溶液, 常温条件下分三次滴加共3倍当量的40 %过氧乙酸的乙酸溶液, 每隔6 h滴加一次, 每次12.8 g, 每次滴加前补加5 g碳酸氢钠, 全部滴加完后再反应12 h, 薄层色谱(TLC)显示反应结束。用二氯甲烷(2×100 mL)萃取, 合并有机相, 有机相用饱和碳酸氢钠水溶液和饱和食盐水洗两次, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 得白色固体10.9 g, 收率98.6%, m.p.286.5~288 ℃(文献值: 286~289 ℃[11]。) 1H NMR (500 MHz, CDCl3) δ: 4.37~4.51(m, 1H), 3.02 (t, J=5.8 Hz, 1H), 1.64~2.06 (m, 12H); 13C NMR (126 MHz, CDCl3) δ: 179.00, 73.19, 41.24, 35.76, 33.78, 30.95, 25.83; FT-IR (ν/cm-1) : 2914, 2852, 1441 (C—H), 1708 (C=O), 1165 (C—O)。Anal.Calcd for C10H14O2: C, 72.26; H, 8.49; Found: C, 72.47; H, 8.68。
2.3.2 4-羟基金刚烷-2-酮(2)的合成向100 mL单口烧瓶中加入11 g(66 mmol)化合物1和50 mL浓硫酸, 60 ℃反应5 h, 反应结束后将反应液倒入100 mL冰水中, 过滤, 虑饼用水洗2次, 虑液先用二氯甲烷(2×50 mL)萃取, 回收3.5 g原料1, 再加入100 mL水, 110 ℃回流8 h。用二氯甲烷(3×100 mL)萃取, 合并有机相, 有机相用饱和碳酸氢钠水溶液洗两次, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 粗产品用二氯甲烷/正己烷重结晶, 得白色颗粒状晶体5.8 g, 产率77.3%(以反应的化合物1计), m.p.317.2~319.4 ℃(文献值: 316~320 ℃[11])。1H NMR (500 MHz, CDCl3) δ: 4.29 (dd, J=5.1, 3.1 Hz, 1H), 2.65 (s, 1H), 2.51 (s, 1H), 2.42 (d, J=13.0 Hz, 1H), 1.89~2.12 (m, 10H); 13C NMR (126 MHz, CDCl3) δ: 217.07, 78.19, 54.47, 46.59, 38.90, 37.70, 35.21, 33.71, 33.18, 26.29; FT-IR(ν/cm-1) : 3390 (O—H), 2916, 2856, 1450 (C—H), 1698 (C=O), 1046 (C—O)。Anal.Calcd for C10H14O2: C, 72.26; H, 8.49; Found: C, 72.3; H, 8.69。
2.3.3 4, 4-乙撑二氧基金刚烷-2-酮(4)的合成向250 mL单口烧瓶中加入5.7 g(34 mmol)化合物2、1.62 g(8.6 mmol)对甲苯磺酸、2.83 mL(51 mmol)乙二醇和175 mL甲苯, 加热至125 ℃回流, 甲苯共沸除水, 反应3 h, TLC显示反应结束。减压蒸馏除甲苯得深棕色油状液体, 油状液体用100 mL二氯甲烷溶解, 有机相用饱和碳酸氢钠水溶液和饱和食盐水洗两次, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 得7.1 g浅棕色油状液体, 待用。
向250 mL单口烧瓶中加入16.63 g(27 mmol)碾磨过的过硫酸氢钾复盐和80 mL乙酸乙酯, 常温搅拌2 h, 再将0.207 g(0.68 mmol)邻碘苯磺酸钠、以上所得的7.1 g油状液体和34 g无水硫酸钠加入反应瓶中, 70 ℃反应6 h。过滤, 虑饼用乙酸乙酯洗涤, 减压蒸馏除溶剂得6.95 g黄色油状液体, 采用柱层析分离得无色液体6.5 g, 两步总收率为92.9%。1H NMR (500 MHz, CDCl3) δ: 3.81~ 4.04 (m, 4H), 2.53 (d, J=27.5 Hz, 2H), 2.34~2.09 (m, 2H), 1.76~2.05 (m, 8H); 13C NMR (126 MHz, CDCl3) δ: 214.07, 111.86, 64.85, 64.65, 56.14, 45.44, 38.85, 37.40, 36.26, 33.73, 32.06, 26.22; FT-IR(ν/cm-1) : 2931, 2860, 1451 (C—H), 1713 (C=O), 1119 (C—O)。Anal.Calcd for C12H16O3: C, 69.21; H, 7.74; Found: C, 69.05; H, 7.82。
2.3.4 4, 4-乙撑二氧基金刚烷-2-酮肟(5)的合成向250 mL单口烧瓶中加入5 g(24 mmol)化合物4和150 mL乙醇, 搅拌至液体全部溶解, 再加入3.35 g(48 mmol)盐酸羟胺和7.86 g(96 mmol)乙酸钠, 常温反应12 h。减压蒸馏除乙醇得固体, 固体用75 mL二氯甲烷溶解, 有机相用饱和碳酸氢钠水溶液洗两次, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 粗产品用乙醇重结晶, 得白色颗粒状晶体4.92 g, 收率91.8 %, m.p.115.2~117.8 ℃(文献值: 116~120 ℃[5])。1H NMR (500 MHz, CDCl3) δ: 8.19 (s, 1H), 3.92~4.08 (m, 4H), 3.61 (s, 1H), 2.52 (s, 1H), 2.09 (ddd, J=14.5, 7.5, 2.6 Hz, 3H), 1.75~1.93 (m, 7H). 13C NMR (126 MHz, CDCl3) δ: 164.65, 164.23, 111.09, 110.98, 64.66, 64.49, 44.91, 38.42, 37.57, 36.89, 36.81, 36.55, 36.51, 35.16, 35.03, 34.03, 34.00, 33.92, 32.81, 27.66, 26.66, 26.58. FT-IR(ν/cm-1) : 3209 (O—H), 2915, 2852, 1447 (C—H), 1666 (—C=N—), 1120(C—O)。Anal.Calcd for C12H17NO3: C, 64.55; H, 7.67; N, 6.27; Found: C, 64.62; H, 7.69; N, 6.22。
2.3.5 2, 2-二硝基-4, 4-乙撑二氧基金刚烷(6)的合成向250 mL三口烧瓶中加入3.65 g(16.4 mmol)化合物5和164 mL干燥后的二氯甲烷, 搅拌至固体溶解, 再加入5 g无水硫酸钠和2.94 g(49.2 mmol)尿素, 45 ℃回流, 将5.3 g(49.2 mmol)五氧化二氮溶于20 mL干燥后的二氯甲烷, 在氮气保护下慢慢滴加入反应瓶中, 10 min内滴加完, 开始滴加到反应结束共30 min。将反应液倒入冷的饱和碳酸氢钠水溶液中, 用二氯甲烷(3×75 mL)萃取, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 得黄色油状液体, 采用柱层析分离得2.43 g白色固体, 回收0.76 g 4, 4-乙撑二氧基金刚烷-2-酮(4), 收率52.4%, m.p. 232.6~233.9 ℃。1H NMR (500 MHz, CDCl3) δ: 4.04~4.08 (m, 1H), 3.87~3.96 (m, 2H), 3.80~3.86 (m, 1H), 3.39 (s, 1H), 3.28 (d, J=2.8 Hz, 1H), 2.36-2.43 (m, 1H), 2.24 (dd, J=14.1, 2.9 Hz, 1H), 1.56~2.03 (m, 8H); 13C NMR (126 MHz, CDCl3) δ: 123.13, 109.69, 64.81, 64.34, 41.20, 35.53, 34.03, 33.99, 32.45, 32.23, 30.75, 24.31. FT-IR(ν/cm-1) : 2930, 2861, 1459 (C—H), 1572, 1314 (—NO2), 1131 (C—O)。Anal.Calcd for C12H16N2O6: C, 50.70; H, 5.67; N, 9.85; Found: C, 50.76; H, 5.69; N, 9.81。
2.3.6 4, 4-二硝基金刚烷-2-酮(7)的合成向100 mL的单口烧瓶中加入1.7 g(6 mmol)化合物6和60 mL二氯甲烷, 搅拌至固体溶解, 再加入21 mL浓硫酸, 常温反应1 h。将反应液倒入冰水中, 用二氯甲烷(3×50 mL)萃取, 合并有机相, 有机相用饱和碳酸氢钠水溶液洗两次, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 粗产品用二氯甲烷/正己烷重结晶, 得白色颗粒状晶体1.40 g, 收率97.4%, m.p.245.6~247.2 ℃(文献值: 246~247 ℃[5])。1H NMR (500 MHz, CDCl3):δ 3.82 (s, 1H), 3.32 (dd, J=5.5, 2.7 Hz, 1H), 2.72 (s, 1H), 1.81~2.33 (m, 9H); 13C NMR (126 MHz, CDCl3)δ: 206.66, 123.36, 51.89, 43.87, 40.06, 38.03, 34.24, 33.87, 30.52, 25.62; FT-IR(ν/cm-1) : 2947, 2877, 1449(C—H), 1727 (C=O), 1562, 1315 (—NO2)。Anal.Calcd for C10H12N2O5: C, 50.02; H, 5.04; N, 11.66; Found: C, 50.14; H, 5.06; N, 11.62。
2.3.7 4, 4二硝基金刚烷-2-酮肟(8)的合成向100 mL的单口烧瓶中加入1.4 g(5.4 mmol)化合物7和75 mL乙醇, 搅拌至固体全部溶解, 再加入0.72 g(10.8 mmol)盐酸羟胺和2.88 g(21.6 mol)乙酸钠, 常温反应12 h。减压蒸馏除乙醇得固体, 固体用50 mL二氯甲烷溶解, 有机相用饱和碳酸氢钠水溶液洗两次, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 粗产品用乙醇重结晶, 得白色颗粒状晶体1.24 g, 收率89.9%, m.p.183.1~185.8 ℃(文献值: 182~185 ℃[5])。1H NMR (500 MHz, CDCl3) δ: 7.74 (s, 2H), 4.99 (s, 1H), 4.01 (s, 1H), 3.62 (s, 1H), 3.42 (d, J=19.1 Hz, 2H), 2.69 (s, 1H), 2.00~2.16 (m, 12H), 1.92 (t, J=13.3 Hz, 4H), 1.81 (d, J=13.5 Hz, 2H).13C NMR (126 MHz, CDCl3) δ: 159.19, 158.30, 122.28, 122.20, 41.74, 39.09, 36.80, 36.31, 35.33, 34.81, 34.28, 33.96, 33.87, 33.81, 32.57, 32.33, 29.71, 26.84, 25.60, 25.40. FT-IR (ν/cm-1) : 3278 (O—H), 2932, 2862, 1457 (C—H), 1671 (—C=N—), 1589, 1312 (—NO2)。Anal.Calcd for C10H13N3O5: C, 47.06; H, 5.13; N, 16.46; Found: C, 47.23; H, 5.14; N, 16.39。
2.3.8 2, 2, 4, 4-四硝基金刚烷(9)的合成向250 mL三口烧瓶中加入1.1 g(4.3 mmol)4, 4二硝基金刚烷-2-酮肟(8)和43 mL干燥的二氯甲烷, 搅拌至固体全部溶解, 再加入2.5 g无水硫酸钠和0.77 g(12.9 mmol)尿素, 45 ℃回流, 将1.39 g(12.9 mmol)五氧化二氮溶于10 mL干燥的二氯甲烷中, 在氮气保护下慢慢滴加入反应瓶中, 10 min内滴加完, 开始滴加到反应结束共30 min。将反应液倒入冷的饱和碳酸氢钠溶液中, 用二氯甲烷(3×50 mL)萃取, 收集有机相, 无水硫酸钠干燥, 过滤后减压蒸馏除溶剂, 得黄色油状液体, 采用柱层析分离得0.65 g白色固体, 回收0.36 g 4, 4-二硝基金刚烷-2-酮(7), 收率48.1%, m.p.138.4~139.5 ℃(文献值: 138~140 ℃[5])。1H NMR (500 MHz, CDCl3) δ: 4.67~4.73(m, 1H), 3.40 (d, J=2.5 Hz, 2H), 2.74~2.82 (m, 1H), 2.29~2.35 (m, 3H), 1.79~2.09 (m, 5H); 13C NMR (126 MHz, CDCl3) δ: 122.29, 39.13, 34.55, 32.06, 30.74, 23.34; FT-IR(ν/cm-1) : 2958, 1470 (C—H), 1577, 1305 (—NO2)。Anal.Calcd for C10H12N4O8:C, 37.98; H, 3.82; N, 17.72; Found: C, 38.07; H, 3.75; N, 17.63。
3 结果与讨论 3.1 4, 4-乙撑二氧基金刚烷-2-酮(4)的合成工艺关于4, 4-乙撑二氧基金刚烷-2-酮的合成, Jean-Ho Chu[12]等采用两步合成法, 先将金刚烷酮直接氧化成金刚烷-2, 4-二酮, 再进行羰基的单保护, 此法第一步的产率低, 两步总收率为31%; Faulkner[11, 13]等采用四步合成法, 先通过两步合成化合物2, 再将化合物2保护后氧化合成了4, 4-乙撑二氧基金刚烷-2-酮, 虽然此法比第一种方法多两步, 但此法总收率比第一种方法要高, 总收率为55%。本研究采用第二种方法合成4, 4-乙撑二氧基-2-金刚烷酮, 改进了化合物1和化合物2的合成方法, 并采用了新的氧化方法将化合物3氧化成4, 4-乙撑二氧基金刚烷-2-酮, 总收率提高到70.2%。
3.1.1 4-氧杂三环[4.3.1.13, 8]十一烷-5-酮(1)的合成工艺金刚烷酮通过Baeyer-Villiger反应可以合成化合物1。Faulkner[11]等通过SeO2催化双氧水与金刚烷酮反应合成化合物1, 收率为96%。Takayoshi Hara[14]等通过Sn/TN催化双氧水与金刚烷酮反应合成化合物1, 收率为81%。双氧水与金刚烷酮进行Baeyer-Villiger反应所需催化剂如二氧化硒、负载锡等都很昂贵。本研究采用过氧乙酸在碱性条件下与金刚烷酮反应合成化合物1, 此法不需用到昂贵的催化剂, 后处理也简单, 收率可达98.6%。
3.1.2 4-羟基金刚烷-2-酮(2)的合成金刚烷酮在催化剂存在下通过双氧水直接氧化可合成4-羟基金刚烷-2-酮, 但此法产率只有40%[15]。其中生成的内酯1在酸性条件下进行内酯的重排反应合成, 其反应机理如Scheme 2所示:

|
Scheme 2 Reaction mechanism of acid catalyzed rearrangement to afford 4-hydroxyadamantan-2-one |
Faulkner [11]用50%的硫酸催化化合物1的重排反应合成4-羟基金刚烷-2-酮, 反应不能完全进行, 造成了原料和产物分离较困难, 产率为70%。本研究参考Gilbert[16-17]等采用的方法, 改用浓硫酸催化此反应。使用浓硫酸会生成金刚烷醇单硫酸酯, 此化合物易溶于水, 所以进行水解反应之前, 可用二氯甲烷萃取回收原料1。单硫酸酯在水中加热回流条件下就能进行水解反应生成化合物2, 用二氯甲烷萃取把化合物从水中分离出来, 粗产品重结晶后可得纯品。此法产率有所提高, 可达77.3%。
3.1.3 羟基氧化反应将金刚烷仲碳上羟基氧化成羰基, 文献已报道了多种氧化方法。Kikukawa[18]等以TBA8[{Zn(OH2)(μ3-OH)}2{Zn(OH2)2}2{γ-HSiW10O36}2]·9H2O催化双氧水氧化金刚烷醇合成金刚烷酮, 产率89%; Hara[19]等以[Pd(OH)2]-/PO43-/NiZn催化氧气氧化金刚烷醇合成金刚烷酮, 产率96%。由此可见, 虽然在催化剂催化下双氧水和氧气的氧化产率很高, 但这两种方所需催化剂都难以制得。Numan[13]等采用琼斯氧化法合成4, 4-乙撑二氧基金刚烷-2-酮, 保护和氧化两步的总收率为80%。
本研究参考Uyanik[20]等采用的氧化方法, 用邻碘苯磺酸钠催化Oxone氧化合成4, 4-乙撑二氧基金刚烷-2-酮, 邻碘苯磺酸钠用量为2 mol(每100 mol化合物3)。此法后处理简单, 过滤除固体后将溶剂乙酸乙酯直接蒸除就可以得到产物; 并且此法产率高, 保护和氧化两步总收率可达92.9%。
3.2 基于五氧化二氮绿色硝化剂的偕硝化反应金刚烷上偕二硝基的引入主要有两种方法:第一种是酮肟直接与98%硝酸进行偕硝化反应; 第二种是酮肟经过次卤酸氧化卤化、还原和硝基取代反应生成偕二硝基。Archibald[4]等以2-金刚烷酮肟为原料合成2, 2-二硝基金刚烷, 采用第一种偕硝化方法, 收率为21%, 采用第二种方法三步总收率为46%。Dave[5]等采用的是以上第一种偕硝化的方法, 第一步偕硝化产率为35.3%, 第二步偕硝化的产率为29%。
本研究使用绿色硝化剂五氧化二氮进行偕硝化反应, 第一步产率提高到52.4%, 第二步产率提高到48.1%。以第一步偕硝化反应为模版, 控制化合物5的加入量为2 mmol, 溶剂的加入量为20 mL, 考察反应介质、反应时间、反应温度及物料比对收率的影响。
3.2.1 物料配比的影响在45 ℃下, 以二氯甲烷为溶剂, 反应时间为30 min, 改变N2O5加入量, 考察化合物5与N2O5物料比对收率的影响, 结果见表 1。
| 表 1 物料比对收率的影响 Tab.1 Effect of molar ratio of the materials on yield |
由表 1可见, 随着N2O5量的增加收率呈先升高后降低的趋势, 物料比为1:3时, 产率达最高。由于N2O5在该反应中既作为硝化剂又作为氧化剂, 因此至少加入两倍当量的N2O5, 底物才能反应完全。N2O5在回流条件下易分解, 增加N2O5的量可保证反应的顺利进行, 收率会提高; 当N2O5的量继续增加时, 体系酸性会越强, 产物和原料都更易变成酮, 收率有所降低。因此, 适宜的物料比为n(5):n(N2O5)=1:3。
3.2.2 反应介质的影响在45 ℃下, n(5):n(N2O5)=1:3, 反应时间为30 min, 选取了硝基甲烷、二氯甲烷、乙腈、氯仿为反应介质, 考察反应介质对收率的影响, 结果见表 2。
| 表 2 反应介质对收率的影响 Tab.2 Effect of solvent on yield |
由表 2可见, 二氯甲烷的和氯仿两种氯代烃对反应更有利, 收率高于硝基甲烷和乙腈。这种结果可能是:酮肟和N2O5在二氯甲烷和氯仿中溶解度更好, 使得原料浓度更高, 因此有利于反应进行。硝基甲烷和乙腈都与水互溶, 后处理时反应液加入冰水淬灭后, 造成产物分离更困难, 损失较大。和二氯甲烷相比, 氯仿毒性更大, 所以选择二氯甲烷为后续实验的溶剂。
3.2.3 反应时间的影响在45 ℃下, 以二氯甲烷为溶剂, n(5):n(N2O5) =1:3, 考察反应时间对收率的影响, 具体结果见表 3。
| 表 3 反应时间对收率的影响 Tab.3 Effect of reaction time on yield |
由表 3可见, 随着反应时间的增加, 收率呈先升高后降低的趋势, 30 min时收率最高。由于此反应过程中会生成硝酸, 而产物长期处于酸性条件下不稳定, 会发生分解生成酮, 因此反应时间过长就可能导致产物分解, 收率降低。因此, 适宜的反应时间为30 min。
3.2.4 反应温度的影响以二氯甲烷为溶剂, n(5):n(N2O5)=1:3, 反应时间为30 min, 考察反应温度对收率的影响, 结果见表 4。
| 表 4 反应温度对收率的影响 Tab.4 Effect of reaction temperature on yield |
由表 4可见, 反应温度低时, 硝化体系硝化能力降低, 反应速率慢, 收率较低; 随着温度的升高, 硝化能力增强, 收率逐渐提高; 继续升高温度, N2O5分解加剧, 参与反应的N2O5量减少, 收率有所下降。因此, 适宜的反应温度为45 ℃。
3.2.5 正交实验综合上述单因素试验, 以二氯甲烷为溶剂, 选取物料配比A [n(5):n(N2O5)]、反应时间B(min)、反应温度C(℃)三因素, 每个因素各取三水平(见表 5)。采用L9(33)正交表, 结果见表 6。
| 表 5 因素水平表 Tab.5 Table of factors and levels |
| 表 6 正交试验结果 Tab.6 Results of orthogonally designed tests |
由表 6可见, 极值R3>R2>R1, 因此三个因素对收率影响的主次顺序为: B(反应温度)> C(反应时间)> A(物料配比), 即反应温度对收率影响最大, 反应时间次之, 物料配比影响最小。物料配比A因素列: K2>K3>K1; 反应时间B因素列: K2>K3>K1; 反应温度C因素列: K3>K2>K1, 试验指标是收率, 指标越大越好, 所以挑选每个因素列中K值最大对应的水平, 因此, 最优水平组合为A2B2C3, 即物料配比n(5):n(N2O5) =1:3, 反应时间30 min, 反应温度45 ℃。
3.2.6 优化条件下稳定性实验根据正交试验结果, 以二氯甲烷为溶剂, n(5):n(N2O5)=1:3, 反应时间为30 min, 反应温度为45 ℃优化条件下, 进行平行实验。结果列于表 7。
| 表 7 优化条件下稳定性实验 Tab.7 Stability tests under optimized conditions |
由表 7可见, 正交试验优化的条件下, 偕硝化反应的平均收率为52.3%, 与以上正交试验结果保持一致。
4 结论(1) 以金刚烷酮为原料, 经过Baeyer-Villiger反应、内酯重排、缩酮化、氧化、脱保护、两步偕硝化等反应合成了2, 2, 4, 4-四硝基金刚烷, 总收率为14.2%, 是文献报道结果的2.9倍, 并利用核磁共振、红外、元素分析等对产物进行了表征。
(2) 改进了重要中间体4, 4-乙撑二氧基金刚烷-2-酮的合成方法, 总收率较文献提高了15.2%。
(3) 改用绿色硝化剂五氧化二氮进行偕硝化反应, 并优化了偕硝化反应的工艺, 确定了适宜的反应条件:溶剂为二氯甲烷, n(5):n(N2O5)=1:3, 反应温度为45℃, 反应时间为30 min, 收率为52.3%。
| [1] |
许晓娟, 肖鹤鸣, 居学海, 等. 多硝基金刚烷红外光谱和热力学性质的理论研究[J].
含能材料, 2005, 13(1): 40-44. XU Xiao-juan, XIAO He-min, JU xue-hai, et al. Theoretical study on the vibrational spectra, thermodynamic properties for polynitroadamantanes[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2005, 13(1): 40-44. |
| [2] |
盛涤伦. 新一代炸药——多硝基笼状化合物的研究综述[J].
火工品, 1995(4): 34-38. SHENG Di-lun. A novel class of explosives-the summarization of study of the polynitro cage compounds[J]. Initiators & Pyrotechnics, 1995(4): 34-38. |
| [3] |
Sollott G P, Gilbert E E. A facile route to 1, 3, 5, 7-tetraaminoadamantane: synthesis of 1, 3, 5, 7-tetranitroadamantane[J].
J Org Chem, 1980, 45: 5405-5408. DOI:10.1021/jo01314a051 |
| [4] |
Archibald T G, Baum K. Synthesis of polynitroadamantanes: oxidations of oximinoadamantanes[J].
J Org Chem, 1988, 53(20): 4646-4649. |
| [5] |
Dave P R, Ferraro M. Synthesis of 2, 2, 4, 4-tetranitroadamantane[J].
J Org Chem, 1990, 55: 4459-4461. DOI:10.1021/jo00301a047 |
| [6] |
Dave P R, Ferraro L. Composition 2, 2, 4, 4, 6, 6-hexanitroadamantane: US 5202508[P], 1993.
|
| [7] |
Theodore A, Lida Q. Synthesis of 1, 2, 2-trinitroadamantane[J].
J Org Chem, 1995, 60: 1895-1896. DOI:10.1021/jo00111a062 |
| [8] |
许晓娟. 有机笼状高能量密度材料(HEDM)的分子设计和配方设计初探[D]. 南京: 南京理工大学, 2006.
XU Xiao-juan. Primary investigations on the molecular and formulation design of organic cage high energy density materials(HEDM) [D]. Nanjing: Nanjing University of Science and Technolog y, 2006. http: //cdmd. cnki. com. cn/Article/CDMD-10288-2008176253. htm |
| [9] |
何志勇, 罗军, 吕春绪, 等. N2O5硝解DPT制备HMX[J].
火炸药学报, 2010, 33(2): 1-4. HE Zhi-yong, LUO Jun, Lü Chun-xu, et al. Synthesis of HMX from DPT by green nitrolysis with dinitrogen[J]. Chinese Journal of Explosives and Propellants, 2010, 33(2): 1-4. |
| [10] |
Dolenc D, Plesnical B. Abstraction of iodine from aromatic iodides by alkyl radicals: steric and electronic effects[J].
J Org Chem, 2006, 71: 8028 DOI:10.1021/jo061125a |
| [11] |
Faulkner D, McKervey M A. The π-route to substituted adamantanes[J].
J Chem Soc, 1971, 93(16): 3906-3910. |
| [12] |
Jean-Ho Chu, Wan-Sheung Li, Ito Chao, et al. Face selectivity in the reactions of 2, 4-disubstituted adamantanes and their modification by in β-Clusionin solutions[J].
Tetrahedron, 2004, 60: 9493-9501. DOI:10.1016/j.tet.2004.07.075 |
| [13] |
Numan H, Wynberg H. Deuterium and the octant rule for ketones: syntheses and circular dichroism data of chiral 4-deuterioadamantan-2-ones[J].
J Org Chem, 1978, 43(11): 2232-2236. DOI:10.1021/jo00405a030 |
| [14] |
Hara T, Hatakeyama M, Kim A, et al. Preparation of clay-supported Sn catalysts and application to Baeyer-Villiger oxidation[J].
Green Chem, 2012, 14: 771-777. DOI:10.1039/c2gc16437j |
| [15] |
Tejeda D C, Moreno A L, Bermejo F A. Non-hemeiron catalysis in C=C, C-C and CH2 oxidation reactions: oxidative transformations on terpenoids catalyzed by Fe(bpmen)(OTf)2[J].
Tetrahedron, 2013, 69: 2977-2986. DOI:10.1016/j.tet.2013.02.013 |
| [16] |
Gilbert E E. The synthesis of adamantane-2, 4-dione[J].
Synth Commun, 1985, 15(1): 53-56. DOI:10.1080/00397918508063778 |
| [17] |
Shen D M, Langhorne P. Synthesis of adamantane-2, 4-dione: US 5298666[P], 1994.
|
| [18] |
Kikukawa Y, Yamaguchi K, Mizun N. Zinc(Ⅱ) containing γ-keggin sandwich-type silicotungstate: synthesis in organic media and oxidation catalysis[J].
Angew Chem Int Ed, 2010, 49: 6096-6100. DOI:10.1002/anie.201001468 |
| [19] |
Hara T, Sawada J, Nakamura Y, et al. Ananionic D-valine-palladium (Ⅱ) complex supported on a hydroxy double salt with a Brϕnsted basic phosphate anion: application for a heterogeneous catalyst toward aerobic alcohol oxidation[J].
Catal Sci Technol, 2011(1): 1376-1382. |
| [20] |
Uyanik M, Akakura M, Ishihara K. 2-Iodoxybenzenesulfonic acidasan extremely active catalyst for the selective oxidation of alcohols to aldehydes, ketones, carboxylic acids and enones with oxone[J].
J Am Chem Soc, 2009, 131: 251-262. DOI:10.1021/ja807110n |
2, 2, 4, 4-Tetranitroadamantane was synthesized from adamantan-2-one via a nine-step process including Baeyer-Villiger oxidation, lactone rearrangement, ketalization, oxidation and nitration.