




2. Department of Materials Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, China
2. 西南科技大学材料科学与工程学院,四川 绵阳 621010
Over the past decades, more and more researchers have interested the development of the smart energetic materials with high detonation properties, low sensitivities and good thermal stabilities [1-3]. However, these properties are often contradictory to each other [4], making the development of novel energetic materials becomes a difficult and challenging task. Diazole (imidazole and pyrazole) based compounds have attracted a great deal of attention because of their high heats of formation (HOFs) and good thermal stabilities [5-8]. For example, 2, 4-dinitroimidazole [9-10], 2, 4, 5-trinitroimidazole [11-12] and 3, 4, 5-trinitropyrazole [13-15] have been synthesized and investigated to be favorable explosives. It is known that the hydrogen attached to N1 atom is quite acidic and labile in nitrodiazole based compounds. When the nitro (—NO2) or azido (—N3) group is attached to N1 atom of nitrodiazole based compounds, the HOFs and densities increase, whereas they exhibit poor thermal stabilities and safety characteristics. In contrast, the presence of NH2 group will enhance the stabilities, HOFs, densities and detonation performance [16]. Moreover, the amino group can undergo further functionalization, such as nitration [17-18], diazotization [19] to provide versatile energetic materials.
As is well known, the dissociation energy of the weakest bond plays an important role in the initiation of detonation. Moreover, the stabilities of energetic materials have frequently been related to energy of the weakest bond [20]. Besides, the HOF is one of the most crucial thermodynamic quantities and it is usually taken as indicator of the "energy content" of an energetic compound [21-22]. However, to the best of our knowledge, for N-aminopolynitrodiazoles, no data are available for the HOFs in solid state as well as bond dissociation energies (BDEs).
Thus, this work theoretically investigated the HOFs in condensed phase, densities, detonation properties, frontier energy gaps (ΔE), BDEs, stabilities and sensitivities of the six N-aminopolynitrodiazoles by density functional theory (DFT) method at B3LYP/aug-cc-pVDZ level. Meanwhile, two known explosives 2, 4-dinitroimidazole (2, 4-DNI) and 3, 4-dinitropyrazole (3, 4-DNP) were computed using the same method, which were used to prove the results.
2 Methods and computational detailsThe six N-aminopolynitrodiazoles were optimized at B3LYP/aug-cc-pVDZ level using the Gaussian 09 quantum chemical package [23]. The optimized structures were positively identified to be true local minima of the potential energy surfaces with no imaginary frequencies by frequency calculations.
Detonation velocity and pressure were evaluated by the widely used empirical Kamlet-Jacobs equations [24]:
| $ D = 1.01{(N{\overline M ^{1/2}}{Q^{1/2}})^{1/2}}(1 + 1.3\rho ){\rm{ }} $ | (1) |
| $ p = 1.558N\overline M {^{1/2}}{Q^{1/2}}{\rho ^2} $ | (2) |
Where D is the detonation velocity, km·s-1; p is the detonation pressure, GPa; N is the number of moles of gaseous detonation products per gram of explosive, mol·g-1; M is the average molecular weight of the gaseous products, g·mol-1; ρ is the initial density of the explosive, g·cm-3; and Q is the chemical energy of the detonation reaction, J·g-1.
In this study, the densities of the title compounds were calculated using the following equation suggested by Politzer et al.[25].
| $ \rho = \alpha^\prime(\frac{{{M_{{\rm{mol}}}}}}{{{V_{\rm{m}}}}}) + \beta^\prime \left( {v\sigma _{{\rm{tot}}}^2} \right) + \gamma^\prime {\rm{ }} $ | (3) |
Where Mmol is the molecular mass, g·mol-1; Vm is the volume defined as the space inside a counter of electron density of 0.001 e/Bohr3, Å3·mol-1; α′, β′ and γ′are regression coefficients and their values are taken from reference [25].
The Q can be evaluated from the HOFs of the products and reactants. The standard gas phase HOFs (ΔHf (g, 298 K)) were calculated using atomization approach as following [26-28]:
| $ \Delta {H_{\rm{f}}}\left( {M, {\rm{ }}0{\rm{K}}} \right) = {\rm{ }}\sum\limits_{{\rm{atom}}} x \Delta {H_{\rm{f}}}\left( {{\rm{X}}, {\rm{ }}0{\rm{K}}} \right) - [\sum\limits_{{\rm{atom}}} x {\varepsilon _0}\left( {\rm{X}} \right)- {\varepsilon _0}\left( {\rm{M}} \right)- {\varepsilon _\text{ZPE}}\left( {\rm{M}} \right)] $ | (4) |
| $ \begin{array}{l} \Delta {H_{\rm{f}}}\left( {{\rm{g}}, {\rm{ }}298\,\,\,\,{\rm{ K}}} \right){\rm{ }} = {\rm{ }}\Delta {H_{\rm{f}}}(\mathit{M}, {\rm{ }}0{\rm{K}}) + {\rm{ }}\sum\limits_{{\rm{atom}}} x \left( {{H_{\rm{X}}}\left( {0{\rm{K}}} \right){H_{\rm{X}}}\left( {298\,\,\,\,{\rm{ K}}} \right)} \right){\rm{ }} + \\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;{\rm{ }}\left( {{H_{\rm{M}}}\left( {298\,\,\,\,{\rm{ K}}} \right){H_{\rm{M}}}\left( {0{\rm{K}}} \right){\rm{ }}} \right) \end{array} $ | (5) |
Where M denotes the molecule, x is the number of the atom X in M, ε0(M) is the total energy of the molecule, εZPE(M) is its zero-point energy, and (HX (0K)-HX(298 K)) stands for enthalpy corrections for atomic elements, which can be found from reference [29]
However, in the case of energetic compounds, the HOFs in solid state of the designed compounds are needed. They can be estimated using Eq. (6) and (7) developed by Politzer et al. [30-31].
| $ \Delta {H_{\rm{f}}}\left( {{\rm{solid}}, {\rm{ }}298\,\,\,\,{\rm{ K}}} \right) = \Delta {H_{\rm{f}}}\left( {{\rm{gas}}, {\rm{ }}298\,\,\,\,{\rm{ K}}} \right) - \Delta {H_{{\rm{sub}}}}\left( {298\,\,\,\,{\rm{ K}}} \right){\rm{ }} $ | (6) |
| $ \Delta {H_{{\rm{sub}}}}\left( {298\,\,\,\,{\rm{ K}}} \right) = \alpha {({A_{\rm{S}}})^2} + \beta \nu \sigma _{{\rm{tot}}}^2{)^{0.5}} + \gamma $ | (7) |
Where ΔHsub is the sublimation enthalpy, kJ·mol-1. The values of coefficients α, β and γ are taken from reference [30]. As is the molecular surface area, Å2; ν is a measure of the degree of balance between positive and negative potential on the molecular surface, and σtot2 is the total variance of the surface potential, (kJ·mol-1)2.
The strength of the bond and relative stability can be evaluated by the BDE, which means the difference between the energy of a molecule and those of the radicals produced when a bond of this molecule is broken. The energy required for homolytic bond cleavage at 298 K and 1 atm corresponds to the enthalpy of reaction. This has been frequently used as a measure of the thermal stability of the compounds. The BDE of the trigger bond can be given in terms of Eq. (8) [32].
| $ BDE({\rm{R}} - {\rm{X}}) = E\left( {\rm{R}} \right) + E\left( {\rm{X}} \right) - E\left( {{\rm{R}} - {\rm{X}}} \right) $ | (8) |
Where E is the total energy, kJ·mol-1; R-X is the designed molecule. R and X are the radicals produced by dissociation of R-X.
The BDE with zero-point energy (ZPE) correction can be calculated by Eq.(9)[33]
| $ BD{E_{{\rm{ZPE}}}}\left( {{\rm{R}} - {\rm{X}}} \right) = BDE\left( {{\rm{R}} - {\rm{X}}} \right) + \Delta ZPE $ | (9) |
Where ΔZPE is the difference between the ZPE of the products and reactants, kJ·mol-1.
The impact sensitivities can be measured by the characteristic drop height (H50). Here, the H50 of the title compounds can be calculated from the following equation [34]:
| $ \begin{array}{l} {\rm{log}}{H_{50}} = {\rm{ }}(46.2923a + 35.6305b - 7.7005c + 7.9425d + \\ \;\;\;\;\;\;\;\;\;\;\;\;\;{\rm{ }}44.4167{n_{( - {\rm{CNC}} - )}} + 102.2749{n_{( - {\rm{CNNC}} - )}})/{M_{\rm{w}}} \end{array} $ | (10) |
Where a, b, c and d present the number of carbon atoms, hydrogen atoms, nitrogen atoms and oxygen atoms, respectively. n(—CNC—) and n(—CNNC—) are the number of —CNC— and —CNNC— moieties in the aromatic ring. Mw means the average molecular mass, g·mol-1.
3 Results and discussion 3.1 Optimized structuresThe structures of the title compounds are optimized at the DFT-B3LYP/ aug-cc-pVDZ level and the basic structures and the atom numbering schemes are presented in Fig. 1. Optimized bond lengths of the title compounds are listed in Table 1 and corresponding dihedral angles are listed in Table 2. For these compounds, the lengths of C—N (1.313~1.469 Å) are much smaller than normal C—N single bond (1.49 Å)[35]. Bond lengths of C—C bonds are in range from 1.386 Å to 1.414 Å and are also shorter than that of normal C—C single bond (1.54 Å)[36], which can be attributed to the presence of hyperconjugation in the whole molecule. From Table 2, it can be found that the dihedral angles in diazole rings are almost zero (< ±1°) and five atoms in the rings can be considered as nearly coplanar in the title molecules.

|
Fig.1 Molecular frameworks and the atomic numbering of the title compounds, with trigger linkages (C—NO2) encircled in red; the list of values besides each structure shows the trigger bond length (in Å), nitro group charge (in e) and midpoint electrostatic potential from top to bottom, respectively. |
| Tab.1 Selected bond lengths (Å) of the title compounds computed at B3LYP/ aug-cc-pVDZ level |
| Tab.2 Dihedral angles of the title compounds computed at B3LYP/ aug-cc-pVDZ level |
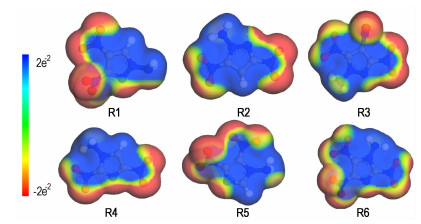
The optimized structures computed at the B3LYP/aug-cc-pVDZ level were employed to calculate the molecular electrostatic potential surface (MESP) using Materials Studio 6.0 package [37]. The MESP of title molecules are given in Fig. 2. Overall, the positive charges are mainly inside, which are stronger and larger in area than that of the negative spatial domains. This is consistent with the views of Klapötke et al. [38] for the energetic compounds.

|
Fig.2 Molecular electrostatic potential surface of six compounds (Red and blue surfaces represent negative and positive regions, respectively, with colors representing values between -2e-2 and 2e-2 Hartree) |
The molecular frontier orbital energies and energy gaps are listed in Table 3. The frontier energy gap (ΔE=ELUMO -EHOMO) between the Highest Occupied molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) can be used to determine the chemical stability of a molecule due to intramolecular charge transfer [39-40]. In general, the smaller the ΔE is, the easier the electron transfer and the lower the molecular chemical stability is. From the ΔE values of the listed compounds, it is noted that the ΔE value of R1 is the largest, and that of R3 is the smallest, which indicates that the former is more stable than the latter. It can be seen that the values decrease with the introduction of —NO2 groups from two to three in the diazole ring. The decreasing order of stability of the compounds is as follows: R1 > R2 > R4 > R5 > R6 > R3.
| Tab.3 The values of ELUMO, EHOMO and ΔE of title molecules computed at the B3LYP/aug-cc-pVDZ level |
As is evident in Table 4, the H50 of compounds with molecules R1, R2, R4 and R5 are higher than that of RDX (26 cm[2]), implying that these compounds are less insensitive than RDX. In addition, H50 of compounds with R1, R2, R4 and R5 are higher than those of R3 and R6, which indicates that the H50 decreases with the increasing of number of nitro groups, that is to say, the dinitro compounds are less insensitive than the trinitro compounds.
| Tab.4 Trigger bond, Wiberg bond order, Vmid, Mulliken charges on nitro group (-QNO2), BDE and H50 of title compounds |
The impact sensitivities of model compounds can also be predicted by the Mulliken atomic charge analysis of nitro group [41]. In explosives, C—NO2 and N—NO2 bonds are the weakest and their breaking is often the initial step in the detonation of explosive. The nitro group charge (-QNO2) is calculated by the sum of net Mulliken atomic charges on the nitrogen and oxygen atoms of the nitro group:
| $ \begin{array}{l} - {Q_{{\rm{N}}{{\rm{O}}_{\rm{2}}}}} = {Q_{\rm{N}}} + {Q_{{\rm{O(1)}}}} + {Q_{{\rm{O}}(2)}}\\ {V_{{\rm{mid}}}} = {\rm{ }}\frac{{2({Q_{\rm{C}}} + {Q_{\rm{N}}})}}{{{R_{({\rm{C}}-{\rm{N}})}}}} \end{array} $ |
Where Vmid is the midpoint electrostatic potential, R(C—N) is trigger length, Å; QC, QN , QO(1) and QO(2) are the Mulliken charges on carbon, nitrogen and oxygen atoms, e, respectively.
The computed trigger bond, Wiberg bond order, Vmid and -QNO2 values of the title compounds are presented in Table 4. Generally, higher negative charge the nitro group possesses, lower the electron-attraction ability is, therefore, the nitro compound is more stable. Variations of the calculated -QNO2 values range from 0.529 e to 0.702 e. Compared with the -QNO2 values of TNT (0.249 e), FOX-7 (0.365 e), RDX (0.134 e), HMX (0.112 e), TNAZ (0.114 e), LLM-105 (0.292 e) and NTO (0.264 e) [41-42], the title compounds appear to be insensitive. According to the Vmid values in Table 4, all molecules may -exhibit lower sensitivities than (TNT) (0.25) and (TATB) (0.42)[42]. The strength of trigger bond can be characterized by Wiberg bond order. The smaller the Wiberg bond order is, the weaker the bond will be and thus the molecule is less stable. These impact sensitivities may attribute to the presence of π-excessive aromatic heterocyclic ring, delocalization of π-electrons and the existence of intra or intermolecular hydrogen bonds.
3.4 BDEIt is known that the BDE of the trigger bond can provide information about the stability of a compound. As a whole, the smaller the BDE is, the less the molecular stability will be. The BDEs have been predicted for the title compounds by breaking the weakest bonds (R—NO2), shown in Table 4. As can be seen from Table 4, the BDEs of all the compounds are in the range of 238.94 kJ·mol-1 to 278.53 kJ·mol-1, and BDEs of the title molecules are higher than those of RDX (152.09 kJ·mol-1) and HMX (166.48 kJ·mol-1)[43], which implies that they have better thermal stabilities. It could also be noted that the BDEs decrease as the number of nitro group increases.
3.5 HOF and DensitiesTable 5 summarizes the ΔHf (g), ΔHf (s) and the related data of AS, vσtot2 and ΔHsub. It is noted from Table 5, all the title compounds show very high positive HOFs (ΔHf (g), 322.62 kJ·mol-1 to 432.39 kJ·mol-1, ΔHf (s), 208.52 kJ·mol-1 to 314.68 kJ·mol-1), and are far higher than those of RDX(ΔHf (s), 191.63 kJ·mol-1) and HMX(ΔHf (g), 258.15 kJ·mol-1). With the increase in the number of the nitro groups, the HOFs always increase. Table 5 also shows that the HOFs are slightly affected by the relative position of the nitro groups. The title compounds possess higher HOFs due to the contributions of N-N bonds as compared with that of the corresponding compounds 2, 4-DNI and 3, 4-DNP.
| Tab.5 Heats of formation (HOFs) and the related parameters of AS, νσtot2 and ΔHsub of the title compounds and some referred compounds |
A convenient method was used to predict the densities of the title compounds proposed by Politzer et al. [25]. The densities are listed in Table 6. The predicted densities of 2, 4-DNI and 3, 4-DNP agree well with experimental values (1.77 g·cm-3 and 1.81 g·cm-3), which indicates this method is reliable. From Table 6, the values have been found to be in range from 1.71 g·cm-3 to 1.82 g·cm-3. The compounds R3 and R6 have equal density 1.82 g·cm-3, which is comparable to the experimental density of RDX.
| Tab.6 Predicted densities, molar volume (Vmol) and detonation performance of the title compounds along with these of experimental values of 2, 4-DNI, 3, 4-DNP, RDX and HMX |
Several empirical or semi-empirical methods have been applied to estimate these parameters [24, 45-46]. Kamlet-Jacobs equations have been proved to be reliable [47-48]. Table 6 shows calculated detonation velocity (D) and detonation pressure (p) of the title compounds along with experimental and calculated values of 2, 4-DNI and 3, 4-DNP. The calculated D and p of R1-R6 are 8.23-8.99 km·s-1 and 29.16-36.12 GPa, respectively. It is observed that R3 (8.99 km·s-1, 36.12 GPa) and R6(8.92 km·s-1, 35.56 GPa) show better properties in the series due to the better densities and higher HOFs. Calculation results indicate that the values of R3 (8.99 km·s-1, 36.12 GPa) and R6 (8.92 km·s-1, 35.56 GPa) appear to be superior to that of RDX (8.75 km·s-1, 34.70 GPa) and comparable to that of HMX (9.1 km·s-1, 39.00 GPa). Additionally, the detonation performance of compounds with molecules R1, R2, R4 and R5 is similarly to that of RDX. In a word, compounds with R1-R6 have comparable detonation performances to RDX and HMX.
4 ConclusionIn this paper, density functional theory was employed to study the electronic structures, thermal stabilities, heats of formation, densities and explosive properties of N-aminopolynitrodiazoles. Results are as follows:
(1) The ΔE of the title molecules decrease as the number of nitro groups increases. Based on atomization approach, standard gas phase HOFs of the title compounds were predicted. The solid state HOFs were estimated in the framework of the Politzer model. Results show that all title molecules possess high positive HOFs (ΔHf (g), 322.62 kJ·mol-1 to 432.39 kJ·mol-1, ΔHf (s), 208.52 kJ·mol-1 to 314.68 kJ·mol-1). The HOFs are slightly affected by the relative position of the nitro groups.
(2) The analysis of the bond dissociation energy of the weakest bond (238.94 kJ·mol-1 to 278.53 kJ·mol-1) suggests that the title molecules are with good thermal stabilities. The other analysis of the nitro group charge and H50 shows all the compounds have low impact sensitivities.
(3) The D, p and ρ also increase as the number of nitro groups increases. R3 (8.99 km·s-1, 36.12 GPa) and R6 (8.92 km·s-1, 35.56 GPa) show better detonation properties in the series due to the better densities and higher HOFs. All title molecules show comparable detonation performances (8.23-8.99 km·s-1 and 29.16-36.12 GPa) to RDX (8.75 km·s-1, 34.70 GPa) and HMX (9.1 km·s-1, 39.00 GPa).
| [1] |
Agrawal J P, Hodgson R D. Organic Chemistry of Explosives[M]. John Wiley: Sons, 2007.
|
| [2] |
Pagoria P F, Lee J S, Mitchell A R, et al. A review of energetic materials synthesis[J]. Thermochimica Acta, 2002, 384: 187-204. DOI:10.1016/S0040-6031(01)00805-X |
| [3] |
Agrawal J P. Recent trends in high-energy materials[J]. Progress in Energy and Combustion Science, 1998, 24: 1-30. DOI:10.1016/S0360-1285(97)00015-4 |
| [4] |
Sikder A K, Sikder N. A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications[J]. Journal of Hazardous Materials, 2004, 112: 1-15. DOI:10.1016/j.jhazmat.2004.04.003 |
| [5] |
Zaitsev A A, Dalinger I L, Shevelev S A. Dinitropyrazoles[J]. Russian Chemical Reviews, 2009, 78: 589-627. DOI:10.1070/RC2009v078n07ABEH004015 |
| [6] |
Bracuti A J. Crystal structure of 2, 4-dinitroimidazole (2, 4-DNI)[J]. Journal of Chemical Crystallography, 1995, 25: 625-627. DOI:10.1007/BF01665967 |
| [7] |
Cho J R, Kim K J, Goh E M, et al. Synthesis and characterization of 1-methyl-2, 4, 5-trinitroimidazole (MTNI)[J]. Journal of Hazardous Materials, 2002, 39: 141-147. |
| [8] |
Ravi P, Badgujar D M, Gore G M, et al. Review on melt cast explosives[J]. Propellants Explosives Pyrotechnics, 2011, 36: 393-403. DOI:10.1002/prep.201100047 |
| [9] |
Bhaumik K, Akamanchi K G. 2, 4-Dinitroimidazole: microwave assisted synthesis and use in synthesis of 2, 3-dihydro-6-nitroimidazo[2, 1-b] oxazole analogues with antimycobacterial activity[J]. Journal of Hazardous Materials, 2004, 41: 51-55. |
| [10] |
Bulusu S, Dhamavarapu R, Autera J R, et al. Thermal rearrangement of 1, 4-dinitroimidazole to 2, 4-dinitroimidazole: characterization and investigation of the mechanism by mass spectroscopy and isotope labeling[J]. Journal of Physical Chemistry, 1995, 99: 5009-5015. DOI:10.1021/j100014a022 |
| [11] |
SU Xin-fang, CHENG Xin-lu, GE Su-hong. Theoretical investigation on structure and properties of 2, 4, 5-trinitroimidazole and its three derivatives[J]. Journal of Molecular Structure: Theochem, 2009, 895: 44-51. DOI:10.1016/j.theochem.2008.10.006 |
| [12] |
Katritzky A R, Cundy D J, Chen J. Polyiodoimidazoles and their nitration products[J]. Journal of Energetic Materials, 1993, 11: 345-352. DOI:10.1080/07370659308019716 |
| [13] |
Dalinger I L, Popova G P, Vatsadze I A. Synthesis of 3, 4, 5-trinitropyrazole[J]. Russian Chemical Bulletin, 2009, 58: 2185. DOI:10.1007/s11172-009-0301-2 |
| [14] |
Hervé G, Roussel C, Graindorge H. Selective preparation of 3, 4, 5-Trinitro-1H-pyrazole: a stable all-carbon-nitrated arene[J]. Angewandte Chemie, 2010, 49: 3177-3181. DOI:10.1002/anie.v49:18 |
| [15] |
Ravi P, Reddy C K, Saikia A. Nitrodeiodination of polyiodopyrazoles[J]. Propellants, Explosives, Pyrotechnics, 2012, 37: 167-171. DOI:10.1002/prep.v37.2 |
| [16] |
Larina L, Lopyrev V. Nitroazoles: synthesis, structure and application[M]. Springer: New York, 2009.
|
| [17] |
Joo Y H, Shreeve J M. Energetic mono-, di-, and trisubstituted nitroiminotetrazoles[J]. Angewandte Chemie, 2009, 121: 572-575. DOI:10.1002/ange.v121:3 |
| [18] |
Duddu R, Dave P R, Damavarapu R, et al. Synthesis of N-amino-and N-nitramino-nitroimidazoles[J]. Tetrahedron Lett, 2010, 51: 399-401. DOI:10.1016/j.tetlet.2009.11.046 |
| [19] |
QI Cai, LI Sheng-hua, LI Yu-chuan, et al. A novel stable high-nitrogen energetic material: 4, 4'-azobis(1, 2, 4-triazole)[J]. Journal of Materials Chemistry, 2011, 21: 3221-3225. DOI:10.1039/c0jm02970j |
| [20] |
Rice B M, Sahu S, Owens F J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules[J]. Journal of Molecular Structure: Theochem, 2002, 583: 69-72. DOI:10.1016/S0166-1280(01)00782-5 |
| [21] |
FAN Xiao-wei, Ju Xue-xue. Theoretical studies on four-membered ring compounds with NF2, ONO2, N3, and NO2 groups[J]. Journal of Computional Chemistry, 2008, 29: 505-513. DOI:10.1002/jcc.20809 |
| [22] |
Cobos C J. DFT study of the thermochemistry of gas-phase 1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetraazacyclooctane (β-HMX)[J]. Journal of Molecular Structure: Theochem, 2005, 714: 147-152. DOI:10.1016/j.theochem.2004.09.042 |
| [23] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09. Revision A. 01[CP]. Gaussian Inc. Wallingford CT, 2009.
|
| [24] |
Kamlet M J, Jabcobs S J. Chemistry of detonation. I. a simple method of calculating the detonation properties of C—H—NO explosives[J]. Journal of Chemistry Physical, 1968, 48: 23-35. DOI:10.1063/1.1667908 |
| [25] |
Politzer P, Martinez J, Murray J S, et al. An electrostatic interaction correction for improved crystal density prediction[J]. Molecular Physics, 2009, 107: 2095-2101. DOI:10.1080/00268970903156306 |
| [26] |
ZHOU Yang, LONG Xin-ping, SHU Yue-jie. Theoretical studies on the heats of formation, densities, and detonation properties of substituted s-tetrazine compounds[J]. Journal of Molecular Modeling, 2010, 16: 1021-1027. DOI:10.1007/s00894-009-0605-z |
| [27] |
SHU Yuan-jie, LI Hua-rong, GAO Shi-jie, et al. Theoretical studies on densities, stability and detonation properties of 2D polymeric complexes Cu(DAT)2Cl2 and its new analogues Zn(DAT)2Cl2[J]. Journal of Molecular Modeling, 2013, 19: 1583-1590. DOI:10.1007/s00894-012-1728-1 |
| [28] |
MAN Tian-tian, WANG Kun, ZHANG Jian-guo, et al. Theoretical studies of high-nitrogen-containing energetic compounds based on the s-tetrazine Unit[J]. Central European Journal Energetic Materials, 2013, 10: 171-189. |
| [29] |
Rice B M, Pai S V, Hare J. Predicting heats of formation of energetic materials using quantum mechanical calculations[J]. Combustion and Flame, 1999, 118: 445-458. DOI:10.1016/S0010-2180(99)00008-5 |
| [30] |
Politzer P, Murray J S, Grice M E, et al. Calculation of heats of sublimation and solid phase heats of formation[J]. Molecular Physics, 1997, 91: 923-928. DOI:10.1080/002689797171030 |
| [31] |
Politzer P, Murray J S. Some perspectives on estimating detonation properties of C, H, N, O compounds[J]. Central European Journal Energetic Materials, 2011, 8: 209-220. |
| [32] |
Blanksby S J, Ellison G B. Bond dissocitation energies of organic molecules[J]. Accounts of chemical research, 2003, 36: 255-263. DOI:10.1021/ar020230d |
| [33] |
Lide D R. CRC Handbook of Chemistry and Physics[M]. Boca Raton: CRC Press, 2002.
|
| [34] |
Keshavarz M H, Pouretedal H R, Semnani A. Novel correlation for predicting impact sensitivity of nitrohetero-cyclic energetic molecules[J]. Journal of Hazardous Materials, 2007, 141: 803-807. DOI:10.1016/j.jhazmat.2006.07.046 |
| [35] |
SONG Tian-you, WANG Xing-qiao. Inorganic chemistry[M]. Beijing: Higher Education Press, 1987, 366-367.
|
| [36] |
ZHAO Guo-zheng, LU Ming. Theoretical studies on the crystal structure, thermodynamic properties, detonation performance and thermal stability of cage-tetranitrotetraazabicyclooctane as a novel high energy density compound[J]. Journal of Molecular Modeling, 2013, 19: 57-64. DOI:10.1007/s00894-012-1522-0 |
| [37] |
Materials studio 6. 0[CP]. Accelrys Inc. San Diego, 2011.
|
| [38] |
Hammerl A, Klapötke T M, Nöth H, et al. Synthesis, structure, molecular orbital and valence bond calculations for tetrazoleazide, CHN7[J]. Propellants, Explosives, Pyrotechnics, 2003, 28: 165-173. DOI:10.1002/(ISSN)1521-4087 |
| [39] |
DONG Ju, Guan Lun, Cheng Bu. New route for stabilizing silicon fullerenes[J]. Journal of Physical Chemistry B, 2006, 110: 14619-14622. DOI:10.1021/jp0622839 |
| [40] |
Kietzmann H, Rochow R, Ganteför G, et al. Electronic structure of small fullerenes: evidence for the high stability of C32[J]. Physical Review Letters, 1998, 81: 5378-5381. DOI:10.1103/PhysRevLett.81.5378 |
| [41] |
ZHANG Chao-yang, SHU Yuan-jie, HUANG Yi-gang, et al. Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds[J]. Journal of Physical Chemistry B, 2005, 109: 8978-8982. DOI:10.1021/jp0512309 |
| [42] |
ZHANG Chao-yang. Review of the establishment of the nitro group charge method and its applications[J]. Journal of Hazardous Materials, 2009, 161: 21-28. DOI:10.1016/j.jhazmat.2008.04.001 |
| [43] |
YAN Ting, CHI Wei-Jie, BAI Jing, et al. Computational studies on polynitropurines as potential high energy density materials[J]. Journal of Molecular Modeling, 2013, 19: 2235-242. DOI:10.1007/s00894-013-1764-5 |
| [44] |
SONG Xiao-shu, CHENG Xin-lu, YANG Xiong-dong, et al. Correlation between the bond dissociation energies and impact sensitivities in nitramine and polynitro benzoate molecules with polynitro alkyl groupings[J]. Journal of Hazardous Materials, 2007, 150: 317-321. |
| [45] |
Rothstein L R, Petersen R. Predicting high explosive detonation velocities from their composition and structure[J]. Propellants, Explosives, Pyrotechnics, 1979, 4: 56-60. DOI:10.1002/(ISSN)1521-4087 |
| [46] |
Keshavarz M H, Pouretedal H R. Predicting the detonation velocity of CHNO explosives by a simple method[J]. Propellants, Explosives, Pyrotechnics, 2005, 30: 105-108. DOI:10.1002/(ISSN)1521-4087 |
| [47] |
BAI Jing, CHI Wei-jie, LI Lin-li. Quantum chemical study of aminonitrocyclopentanes as possible high energy density materials (HEDMs)[J]. Central European Journal Energetic Materials, 2013, 10: 467-480. |
| [48] |
WEI Tao, ZHU Wei-hua, ZHANG Xiao-wen, et al. Molecular design of 1, 2, 4, 5-tetrazine-based high-energy density materials[J]. Journal of Physical Chemistry A, 2009, 113: 9404-9412. DOI:10.1021/jp902295v |
Electronic structures, energy gaps and sensitivities of N-aminopolynitrodiazoles were investigated by the density functional theory at B3LYP/aug-cc-pVDZ level. The heat of formation in the solid phase and the density were predicted by the Politzer model. Thermal stabilities were predicted by bond dissociation energies(BDEs), and Kamlet-Jacob equations were employed to predict the detonation performance of the title compounds.