




2. 中北大学材料科学与工程学院, 山西 太原 030051;
3. 山西大学商务学院 信息学院, 山西 太原 030051
2. College of Materials Science and Engineering, North University of China, Taiyuan 030051, China;
3. School of Information, Business College of Shanxi University, Taiyuan 030051, China
近年来, 由Nielsen A T[1]首次合成的六硝基六氮杂异伍兹烷(HNIW, CL-20)由于具有高的能量密度比以及优异的爆速和爆压性能, 引起研究者广泛的关注。然而, 较高的感度制约了它在武器方面的应用, 如何平衡能量与安全性之间的矛盾已成为研究难点。有研究表明, 通过制备共晶可降低CL-20感度, 如CL-20/三硝基甲苯(TNT)[2-3], CL-20/环四亚甲基四硝胺(HMX)[4]和CL-20/苯并三氧化呋咱(BTF)[5], 其中王玉平[6]合成的摩尔比为1:1的CL-20/1, 3-二硝基苯(DNB)共晶兼具了CL-20高能和DNB钝感廉价的优势, 具有优异的使用价值。
目前, 关于CL-20热解机理的研究比较多, Isayev[7]运用从头算分子动力学方法分别研究了单分子和凝聚态CL-20的热解机理, 结果表明二者热解路径并不完全相同, 其中NO2的生成势垒决定了整个反应的热解动力学。张力等[8]运用ReaxFF力场对不同温度和密度条件下CL-20单分子和超晶胞的初始热解路径进行了研究, 结果表明不同密度和温度条件只会对热解的速率有影响, 热解机理不受影响。Patil和Brill[9]通过恒温热失重法和FTIR光谱法研究了CL-20热解动力学, 结果表明低温时N—O2键的断裂反应决定其热解速率, 其他气体产物的浓度比是温度的函数。由于DNB比较钝感, 一般不单独使用, 所以对其热解机理的研究比较少, 对CL-20/DNB共晶热解机理的研究更少。含能材料主要通过在极端条件下发生复杂的化学反应释放能量, 反应速度非常快, 实验研究物质的热解机理存在难度。反应分子动力学模拟可以描述原子水平和飞秒尺度上化学反应的详细信息, 尤其是近期发展起来的ReaxFF反应分子动力学方法, 计算精度与量子力学相当, 速度快, 可以处理上百万级原子的体系, 时间尺度可达飞秒级别, 有助于在原子水平上认识含能材料在极端条件下的反应机理, 得到其能量释放规律[10]。此力场在很多含能材料的冲击起爆、冲击点火和爆轰反应等反应机理的研究方面得到广泛应用[11-23]。本研究基于ReaxFF力场, 运用LAMMPS (large-scale atomic/molecular massively parallel simulator)软件[24]研究了2000, 2500 K以及3000 K条件下CL-20/DNB的热解过程, 通过分析产物随模拟时间的变化得到其热解引发机理, 以期为新材料的设计、开发、合成、运输、起爆、安全性和风险评估等实际问题提供理论指导。
2 ReaxFF力场简介与模拟细节 2.1 ReaxFF力场简介ReaxFF[10]反应力场基于第一性原理, 由Adri van Duin和William A. Goddard提出, 力场势能函数以原子间的键级BOij(Bond Order)为基础。ReaxFF力场的势能表达式如下, 其中主要分为键、角、二面角、共轭、库仑、范德华及调整项等(单位: kJ·mol-1)。
| $ \begin{array}{l} {E_{{\rm{system}}}} = {E_{{\rm{bond}}}} + {E_{{\rm{lp}}}} + {E_{{\rm{over}}}} + {E_{{\rm{under}}}} + {E_{{\rm{val}}}} + {E_{{\rm{pen}}}} + {E_{{\rm{coa}}}} + \\ \;\;\;\;\;\;\;\;\;\;\;\;{E_{{\rm{C2}}}} + {E_{{\rm{triple}}}} + {E_{{\rm{tors}}}} + {E_{{\rm{conj}}}} + {E_{{\rm{H-bond}}}} + {E_{{\rm{vdwaals}}}} + {E_{{\rm{Coulomb}}}} \end{array} $ | (1) |
该系统中各能量项的具体意义为: Ebond表示键能: Elp是和孤对电子相关的能量补偿项; Eover和Eunder分别是过配位的能量矫正项和低配位的能量矫正项; Eval、Ecoa、Epen是价角能量项;EC2是C2修正项; Etriple是三键修正项; Etors和Econj是四体作用项; EH-bond是氢键作用项; Evdwaals和ECoulomb分别是范德华和库伦作用项。其中, Ebond、Eval、Etors是依赖于键级的价键相互作用。
2.2 模拟CL-20/DNB共晶X-ray衍射数据来源于剑桥晶体学数据库(CCDC NO.940129), 由CL-20(图 1a)和DNB(图 1b)按摩尔比为1:1组成, 属于正交晶系, Pbca空间群, 晶格参数a=9.4703 Å, b=13.4589 Å, c=33.620 Å。首先运用MS 6.0(Materials Studio 6.0)软件[25]构建CL-20/DNB的单晶胞模型, 如图 2a所示, 为了避免温度和升温速率波动对反应的影响, 构建了2×2×1的超晶胞模型, 共1664个原子, 如图 2b所示。

|
图 1 CL-20和DNB的分子结构 Fig.1 Molecular structures for CL-20 and DNB |

|
图 2 CL-20/DNB共晶单胞结构和超晶胞结构 Fig.2 The unit cell and supercell structures of CL-20/DNB cocrystal |
运用Lammps软件[24]对CL-20/DNB超晶胞进行优化以获得合理的初始构型, 在300 K下进行500 ps等温等容分子动力学模拟(Canonical ensemble, NVT), 接着采用(iso-thermal-isobaric, NPT)系综对其压力进行10 ps弛豫, 压力为0 GPa。为了验证ReaxFF力场的实用性, 将CL-20/DNB共晶在常温下的晶胞参数与相应实验值[6]进行了对比, 结果如表 1所示, 由表 1可得, ReaxFF力场作用下的模拟值与实验值具有较好的一致性, 说明ReaxFF力场可用于CL-20/DNB共晶的研究。最后分别在2000, 2500 K和3000 K下进行50 ps的NVT分子动力学计算, 选择上述高温条件主要是为了模拟常规高能量密度材料爆轰中柴普曼-柔格(CJ)点的温度波动, 得到不同温度对共晶热解反应的影响, 并且能够兼顾计算机的运算能力与所用时间(在不影响反应路径的条件下提高温度可以加快反应, 节省时间)。采用Berendsen方法[26]对温度和压力进行控制, 耦合参数为100 fs, 使其在设定的范围内波动, 采用周期性边界条件, 步长为0.1 fs, 每隔50 fs记录一次原子轨迹以及键级信息, 键级为0.3。
| 表 1 常温下CL-20/DNB共晶晶胞参数模拟值与实验值 Tab.1 The lattice parameters of the cocrystal of CL-20/DNB calculated by ReaxFF force field compared with the experiment value |
图 3给出了不同温度下体系势能和总物种数随时间的变化情况。从图 3a看不出明显的平衡和诱导期, 体系迅速吸收热量发生初级热解反应, 随着反应的进行, 热解生成一系列中间及最终产物, 并放出大量的热, 因而势能发生明显的下降; 温度越高, 系统达到反应平衡所需时间越短, 热解反应越完全, 生成的产物越多, 势能下降也越多。

|
图 3 不同温度下体系势能以及反应过程中总物种数量随模拟时间的变化 Fig.3 Time evolution of potential energy and total species at various temperatures |
由图 3b可知, 初始晶胞内只有CL-20和DNB两种物质, 几乎在0.01 ps时就有产物生成, 热解温度为2500 K和3000 K时物种总量分别在25 ps和6.9 ps内达到了最大值, 随着反应的进一步发生呈现一定程度的衰减。而在较低的温度(2000 K)时, 前5 ps内物种迅速增加到50种, 而后缓慢增加。由此可知, 温度越高, 共晶分解速率越快, 初期产生的新物种数量越多。
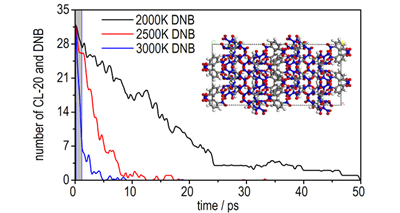
3.2 共晶热解的引发反应机理图 4为2000, 2500 K和3000 K下CL-20及DNB的消耗情况和主要产物的分布。由图 4a可知, 三个温度下CL-20在0.05 ps时均分解完毕, 而DNB消耗较慢, 随着温度的升高其消耗速率显著加快。由图 4b, 图 4c, 图 4d可知, 热解产物主要有NO2、N2、NO、H2O、HNO3、HONO、HON以及CO2等。每个温度下最早生成的产物均为NO2, 其含量在0.05 ps时就达到了最大值, 它是由CL-20中N—O2键和DNB中C—NO2键断裂共同作用的结果, —NO2键的断裂是CL-20/DNB热解的主要引发反应。对于CL-20这种典型的环状硝胺类化合物来说N—NO2键的断裂是其热解的特征引发反应[27], 这与文献[28-30]中的模拟结果一致。此外, 也与Naik和Gore[31]得出N—NO2键的断裂是CL-20热解过程中生成其他小分子产物的前提这一实验结论相互吻合。DNB中的C—NO2键断裂生成NO2是其主要引发反应, 因为C—NO2键是硝基芳香化合物中最弱的键[11]。

|
图 4 不同温度下CL-20及DNB的消耗情况和主要产物随模拟时间的变化 Fig.4 Time evolution of consumption of CL-20 and DNB and main products at various temperatures |
3.2节的分析表明, NO2是共晶热解引发反应所产生, 不同温度下, 各主要产物随时间的变化如图 5所示, 由图 5a可知, NO2数量在达到最大值后会迅速减小, 这是由于次级反应消耗掉大量的NO2, 生成NO、HONO和HNO3等物质。然而在5 ps(2000 K), 6 ps(2500 K)和10 ps(3000 K)左右时NO2数量略有增加, 这主要是由于DNB在此时开始快速分解引起。

|
图 5 主要产物在不同温度下的对比分布曲线 Fig.5 Comparative distribution curves of main products at various temperatures |
NO主要的生成途径有以下几种: (1)两个NO2自由基结合生成N2O4, 接着继续热解生成NO3和NO; (2)对于DNB来说, 由硝基重排形成亚硝酸盐结构(C—NO2→C—ONO), 随后O—NO键断裂也会生成NO, 这与具有相似结构的TNT生成NO的机理相同[11]; (3)由CL-20中N—N键和DNB中C—N键断裂生成的硝基自由基重排布形成亚硝酸盐结构, 通过夺取一个质子生成HONO, 接着分子中N—O键断裂生成NO, 这与Isayev[7]等对凝聚态CL-20热解机理的研究所得结果相吻合; (4)NO2与含碳分子片段生成NO。NO和NO2的分布非常相似, 也是先快速增大然后减少, 大多参与反应生成N2[11], 且温度越高, NO2和NO的消耗速率越快, 生成N2的量越多, 此过程中N2的量一直在不断增多, 如图 5c所示, 也进一步验证了消耗掉的大部分NO2和NO生成了N2。
HONO的主要来源是CL-20和DNB中—NO2键断裂生成的硝基自由基重排布形成亚硝酸盐结构, 通过夺取一个质子所生成, 如图 5d所示随着反应的进行HONO的数量先增多后减少, 主要是由于HONO热解消耗引起。
由图 5e可以看出H2O在整个热解过程中数量先增加然后基本保持不变, 温度越高生成速率越快。水的生成路径有: (1)两个分子间发生质子的转移使硝基生成—NOOH, 然后继续夺取一个质子生成—NOOH—H, 接着脱去一分子的H2O; (2)NO2自由基重排生成的ONO自由基取邻近分子中的质子生成HONO, 随着反应的进行, HONO会进一步热解产生HO自由基, HO自由基继续夺取质子生成H2O, 这与反应过程中HONO先快速积累而后减少是一致的; (3)HNO3的热解也可以产生H2O。
由图 5f可以看出CO2数量随着反应的进行不断增多, 且温度越高, CO2生成越早, 在2000 K时一直到16.85 ps才有CO2生成, 而在3000 K时在2.85 ps就有CO2产生, 这是由于C—C键断裂所需要的能量较多, 一般是在环上基团脱去或者相互反应后, 主环上的C—C键才开始断裂, 进而反应生成CO2。温度为3000 K时, 在40 ps后CO2数量基本保持不变, 这与2000 K和2500 K的现象不同, 其可能的原因在于, 一方面高温下随着反应的进行, 生成的一些小分子片段运动速率加快, 发生碰撞继续聚合生成大分子含碳团簇的几率变大, 使得CO2数量减少; 另一方面随着能量的积累, 大的团簇又发生热解, 最终使得总的数量基本保持不变。通过对产物的分析可知, 产物中确实有少部分大的含碳团簇, 这是含能材料爆轰过程中的常见现象, 一些富碳炸药尤为明显[32-35]。
4 结论(1) 随着温度的升高, 系统达到平衡时间越短, 热解反应越完全, 势能下降越多, 最终总产物生成越多。三个温度下CL-20均先于DNB分解完毕, 升高温度DNB的消耗速率以及产物的生成显著加快。CL-20/DNB共晶热解的主要终产物为NO2、NO、N2、H2O、HNO3、HON、HONO、CO2等, 还有一些重要的中间产物如N2O5、N2O4、OH自由基、N2O等。
(2) 热解反应过程中, CL-20分子中N—NO2键和DNB分子中C—NO2键的断裂生成NO2是最主要的引发反应, 接着两个NO2分子结合或者重排生成ONO自由基, 进一步生成HONO、HON、NO、NO3、H2O等, 其中大部分的NO2和NO继续反应生成N2。3000 K时对产物的分析表明在模拟后期会发生小分子片段的聚合生成大分子碳团簇的现象。
(3) 本研究的热解产物和引发机理与前人的一些结论相互吻合, 说明了ReaxFF力场的实用性与正确性。
| [1] |
Nielsen A T, Chan M L, Kraeutle C K, et al.
Polynitropolyazacaged explosives, part 7, NWC TP7200[M]. China Lake: Navel Weapons Center, 1989.
|
| [2] |
杨宗伟, 张艳丽, 李洪珍, 等. CL-20/TNT共晶炸药的制备, 结构与性能[J].
含能材料, 2013, 20(6): 674-679. YANG Zong-wei, ZHANG Yan-li, LI Hong-zhen, et al. Preparation, structure and properties of CL-20/TNT cocrystal[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2013, 20(6): 674-679. |
| [3] |
Bolton O, Matzger A J. Improved stability and smart-material functionality realized in an energetic cocrystal[J].
Angewandte Chemie International Edition, 2011, 50(38): 8960-8963. DOI:10.1002/anie.v50.38 |
| [4] |
Bolton O, Simke L R, Pagoria P F, et al. High power explosive with good sensitivity: a 2:1 cocrystal of CL-20: HMX[J].
Crystal Growth & Design, 2012, 12(9): 4311-4314. |
| [5] |
YANG Zong-wei, LI Hong-zhen, ZHOU Xiao-qing, et al. Characterization and properties of a novel energetic-energetic cocrystal explosive composed of HNIW and BTF[J].
Crystal Growth & Design, 2012, 12(11): 5155-5158. |
| [6] |
WANG Yu-ping, YANG Zong-wei, LI Hong-zhen, et al. A novel cocrystal explosive of HNIW with good comprehensive properties[J].
Propellants, Explosives, Pyrotechnics, 2013, 35: 1-7. |
| [7] |
Olexandr I, Gorb L, Qasim M, et al. Ab initio molecular dynamics study on the initial chemical events in nitramines: thermal decomposition of CL-20[J].
The Journal of Physical Chemistry B, 2008, 112(35): 11005-11013. DOI:10.1021/jp804765m |
| [8] |
张力, 陈朗, 王晨, 等. CL-20初始热分解反应机理的分子动力学计算[J].
火炸药学报, 2012(4): 5-9. ZHANG Li, CHEN Lang, WANG Chen, et al. Mechanism of the initial thermal decomposition of CL-20 via molecular dynamic simulation[J]. Chinese Journal of Explosives & Propellants, 2012(4): 5-9. |
| [9] |
Patil D G, Brill T B. Thermal decomposition of energetic materials 53 kinetics and mechanism of thermolysis of hexanitrohexazaisowurtzitane[J].
Combustion and Flame, 1991, 87(2): 145-151. DOI:10.1016/0010-2180(91)90164-7 |
| [10] |
VanDuin A C T, Dasgupta S, Lorant F, et al. ReaxFF: a reactive force field for hydrocarbons[J].
The Journal of Physical Chemistry A, 2001, 105(41): 9396-9409. DOI:10.1021/jp004368u |
| [11] |
LIU H, DONG X, HE Y H. Reactive molecular dynamics simulations of carbon-containing clusters formation during pyrolysis of TNT[J].
Acta Physico-Chimica Sinica, 2014, 30(2): 232-240. |
| [12] |
Alejandro, Strachan, Edward M, Kober, Adri C T, van Duin, et al. Thermal decomposition of RDX from reactive molecular dynamics[J].
Journal of Chemical Physics, 2005, 122(5): 054502-054502-10. DOI:10.1063/1.1831277 |
| [13] |
Strachan A, vanDuin A C T, Chakraborty D, et al. Shock waves in high-energy materials: the initial chemical events in nitramine RDX[J].
Physical Review Letters, 2003, 91(9): 098301 DOI:10.1103/PhysRevLett.91.098301 |
| [14] |
Zhou T, Liu L, Goddard Ⅲ W A, et al. ReaxFF reactive molecular dynamics on silicon pentaerythritol tetranitrate crystal validates the mechanism for the colossal sensitivity[J].
Physical Chemistry Chemical Physics, 2014, 16(43): 23779-23791. DOI:10.1039/C4CP03781B |
| [15] |
周婷婷, 石一丁, 黄风雷. 高压下β-HMX热分解机理的ReaxFF反应分子动力学模拟[J].
物理化学学报, 2012, 28(11): 2605-2615. ZHOU Ting-ting, SHI Yi-ding, HUANG Feng-lei. Thermaldecomposition mechanism of β-HMX under high pressures via ReaxFF reactive molecular dynamics simulations[J]. Acta Physico-Chimica Sinica, 2012, 28(11): 2605-2615. DOI:10.3866/PKU.WHXB201208031 |
| [16] |
Zhou TT, Huang F L. Effects of defects on thermal decomposition of HMX via ReaxFF molecular dynamics simulations[J].
The Journal of Physical Chemistry B, 2010, 115(2): 278-287. |
| [17] |
Zhang L, Zybin S V, van Duin A C T, et al. Carbon cluster formation during thermal decomposition of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations[J].
The Journal of Physical Chemistry A, 2009, 113(40): 10619-10640. DOI:10.1021/jp901353a |
| [18] |
vanDuin A C T, Zeiri Y, Dubnikova F, et al. Atomistic-scale simulations of the initial chemical events in the thermal initiation of triacetonetriperoxide[J].
Journal of the American Chemical Society, 2005, 127(31): 11053-11062. DOI:10.1021/ja052067y |
| [19] |
Han S, vanDuin A C T, Goddard Ⅲ W A, et al. Thermal decomposition of condensed-phase nitromethane from molecular dynamics from ReaxFF reactive dynamics[J].
The Journal of Physical Chemistry B, 2011, 115(20): 6534-6540. DOI:10.1021/jp1104054 |
| [20] |
Rom N, Zybin S V, van Duin A C T, et al. Density-dependent liquid nitromethane decomposition: molecular dynamics simulations based on ReaxFF[J].
The Journal of Physical Chemistry A, 2011, 115(36): 10181-10202. DOI:10.1021/jp202059v |
| [21] |
张力, 陈朗. 高压下固相硝基甲烷分解的分子动力学计算[J].
物理学报, 2013, 62(13): 138201-138201. ZHANG Li, CHEN Lang. The effect of pressure on thermal decomposition of solid nitromethane via MD simulation[J]. Acta Physical Sinica, 2013, 62(13): 138201-138201. DOI:10.7498/aps.62.138201 |
| [22] |
Chenoweth K, vanDuin A C T, Dasgupta S, et al. Initiation mechanisms and kinetics of pyrolysis and combustion of JP-10 hydrocarbon jet fuel[J].
The Journal of Physical Chemistry A, 2009, 113(9): 1740-1746. DOI:10.1021/jp8081479 |
| [23] |
Guo F F G, Guo F, et al. ReaxFF molecular dynamics study of initial mechanism of JP-10 combustion[J].
Combustion Science and Technology, 2012, 184: 1233-1243. DOI:10.1080/00102202.2012.679714 |
| [24] |
Plimpton S J, Large-scale atomic/molecular massively parallel simulator[CP]. Sandia National Laboratories: 2003.
|
| [25] |
Materials Studio 6.0[CP]. Accelrys Software Inc, USA: 2013.
|
| [26] |
Berendsen H J C, Postma J P M, van Gunsteren W F, et al. Molecular dynamics with coupling to an external bath[J].
The Journal of Chemical Physics, 1984, 81(8): 3684-3690. DOI:10.1063/1.448118 |
| [27] |
Oxley J C, Kooh A B, Szekeres R, et al. Mechanisms of nitramine thermolysis[J].
The Journal of Physical Chemistry, 1994, 98(28): 7004-7008. DOI:10.1021/j100079a019 |
| [28] |
张力, 陈朗, 王晨, 等. 不同晶型CL-20热分解反应机理计算分析[J].
爆炸与冲击, 2014, 34(2): 188-194. ZHANG Li, CHEN Lang, WANG Chen, et al. Molecular dynamic simulation on thermal decomposition mechanism of CL-20 with different polymorphs[J]. Explosion and Shock Waves, 2014, 34(2): 188-194. DOI:10.11883/1001-1455(2014)02-0188-07 |
| [29] |
张力, 陈朗, 王晨, 等. 水分子对α相CL-20热分解机理影响的分子动力学研究[J].
物理化学学报, 2013, 29(6): 1145-1153. ZHANG Li, CHEN Lang, WANG Chen, et al. Molecular dynamics study of the effect of H2O on the thermal decomposition of α phase CL-20[J]. Acta Physico-Chimica Sinica, 2013, 29(6): 1145-1153. |
| [30] |
刘海, 李启楷, 何远航. CL20-TNT共晶高温热解的ReaxFF/lg反应力场分子动力学模拟[J].
物理学报, 2013, 62(20): 208202 LIU Hai, LI Qi-kai, HE Yuan-hang. Pyrolysis of CL20-TNT cocrystal from ReaxFF/lg reactive molecular dynamics simulations[J]. Acta Physical Sinica, 2013, 62(20): 208202 DOI:10.7498/aps.62.208202 |
| [31] |
Naik N H, Gore G M, Gandhe B R, et al. Studies on thermal decomposition mechanism of CL-20 by pyrolysis gas chromatography-mass spectrometry (Py-GC/MS)[J].
Journal of Hazardous Materials, 2008, 159(2): 630-635. |
| [32] |
Mironov E V, Petrov E A, Korets A Y. From analysis of the structure of ultrafine diamond to the problem of its formation kinetics[J].
Combustion, Explosion and Shock Waves, 2004, 40(4): 473-476. DOI:10.1023/B:CESW.0000033571.82326.6a |
| [33] |
Chevrot G, Sollier A, Pineau N. Molecular dynamics and kinetic study of carbon coagulation in the release wave of detonation products[J].
The Journal of Chemical Physics, 2012, 136(8): 084506 DOI:10.1063/1.3686750 |
| [34] |
Ten K A, Aulchenko V M, Lukjanchikov L A, et al. Application of introduced nano-diamonds for the study of carbon condensation during detonation of condensed explosives[J].
Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 2009, 603(1): 102-104. |
| [35] |
Krüger A, Kataoka F, Ozawa M, et al. Unusually tight aggregation in detonation nanodiamond: identification and disintegration[J].
Carbon, 2005, 43(8): 1722-1730. DOI:10.1016/j.carbon.2005.02.020 |
The initial thermal decomposition pathways as well as some important products generating mechanism of hexanitrohexaazaisowurtzitane(CL-20)/1, 3-dinitrobenzene(DNB)cocrystal at high temperatures(2000, 2500 K and 3000 K) were studied by reactive molecular dynamics simulations using ReaxFF force field.