




2. 军械工程学院弹药保障与安全性评估军队重点实验室,石家庄 050003;
3. 华南师范大学化学与环境学院,广州 510006
2. Military Key Laboratory for Ammunition Support and Safety Evaluation, Ordnance Engineering College, Shijiazhuang 050003, China;
3. School of Chemistry and Environment, South China Normal University, Guangzhou 510006, China
硝酸羟胺是羟胺的硝酸盐,由还原组分羟胺和氧化组分硝酸组成[1-2]。硝酸羟胺基推进剂因具有密度大、比冲高、安全、无毒的优越性能而得到人们的关注,广泛的应用于炮弹的发射、火箭的推进、导弹的姿态调控以及微型卫星的轨道调整[3-7]。目前美国军方已研制出硝酸羟胺基发射药LP1846和LP1898,美国宇航局(NASA)路易斯研究中心在军方液体发射药的基础上开发出用于航天推进的硝酸羟胺基单元推进剂[8]就属于硝酸羟胺基推进剂。
硝酸羟胺基推进剂在点火过程中,通常采用电点火的方式,在不同的电极电势下,电极表面附近的推进剂中存在着一个电势降,即有电场存在,这一区域电场的存在对推进剂的影响尚无详细报道。目前针对外电场对含能材料的影响主要集中于火炸药的电火花感度,Keshavarz等[9]研究了硝胺类炸药的电火花感度与热分解活化能之间的关系,阐明了电火花的热效应对硝胺类炸药的影响,但对于电场效应的影响却没有报道;Keshavarz等[10]研究了硝基芳香化合物的电火花感度与分子的碳氢比之间的关系,建立了分子的组分与电火花感度之间的关系,却忽略了电火花对分子结构本身性质的影响;Türker[11]采用密度泛函的方法研究了硝胺类炸药的电火花感度与分子轨道特征之间的关系,揭示了硝胺类炸药电场下的分解与阴离子轨道特性之间的关系,却忽略了分子的其他特性对电火花感度的影响。
在电场中含能材料在进行可能的化学反应之前,有机分子首先被极化或电离产生离子,然后以阴离子或者阳离子的形式来参与化学反应[12-15]。基于此,本工作针对外电场对硝酸羟胺的氢键作用展开研究,在无电场的情况下优化得到硝酸羟胺的稳定构型,在加载电场的情况下研究氢键和结合能所受到的影响,并对电场作用下的能级分布进行研究,分析外电场对硝酸羟胺的稳定性以及化学活性的影响,以期为硝酸羟胺基推进剂电点火过程中的电场效应的研究提供理论依据。
2 理论和计算方法外电场作用下分子体系的哈密顿量H为[15]:
| $ H = {H_0} + {H_{{\mathop{\rm int}} }} $ | (1) |
式中,H0为无外电场时的哈密顿量,Hint为外电场与分子体系相互作用的哈密顿量。当在偶极近似下,外电场F与分子体系的相互作用哈密顿量可以表示为:
| $ {H_{{\mathop{\rm int}} }} = - \mu \cdot F $ | (2) |
式中,μ为分子电偶极距。
NH3OH+和NO3-在没有外电场下的几何构型如图 1a所示,文献[16]采用DFT-B3LYP/6-311++G(d, p)方法对硝酸铵和硝酸羟胺盐进行了密度泛函研究,本工作根据文献[16]理论计算出的几何结构参数与文献[17]中给出的实验观测结果吻合较好,因此采用B3LYP方法在6-311++G(d, p)基组水平上对硝酸羟胺的可能稳定构型进行几何优化,在此基础上对各构型进行振动分析,计算结果中均无虚频,说明得到的分子结构对应于能量的极小值点。对构型进行自然键轨道(NBO)分析以揭示氢键作用的本质,然后对NH3OH+去质子化解离反应进行理论计算(包括构型优化、频率分析和内禀反应坐标计算),以此来研究构型形成过程中的氢转移,并把过渡态与反应物的能量之差作为反应活化能。为了研究外电场对硝酸羟胺的影响,使用“Field”关键字,沿着硝酸羟胺所在平面方向加载加一系列有限的外电场(-0.005~0.005 a.u.),计算氢转移活化能垒、键长、结合能和能级分布随外电场的变化规律。由于硝酸羟胺分子构型形成过程中出现了氢转移,其构型表现出HNO3和NH2OH相互作用的性质,所以在结合能计算时,将硝酸羟胺的总能量减去NH2OH与HNO3的总能量,并经BSSE和ZPE校正得到各硝酸羟胺的结合能,全部计算在Gaussian09[18]软件包中进行。

|
图 1 NH3OH+、NO3-以及硝酸羟胺优化构型 Fig.1 Optimizad configurations of NH3OH+, NO3-, and hydroxylamine nitrate |
| 表 1 NH3OH+、NO3-以及硝酸羟胺优化构型部分几何参数 Tab.1 Part of geometric parameters for optimized configuration of NH3OH+, NO3- and hydroxylamine nitrate |
采用密度泛函B3LYP/6-311++G(d, p)方法对NH3OH+、NO3-和硝酸羟胺的可能构型进行几何优化,优化几何构型如图 1所示,部分几何参数如表 1所示。NH3OH+属于CS点群,NO3-属于D3h点群,构型Ⅱ属于CS点群,构型Ⅲ和Ⅳ属于C1点群。从表 1可以看出,与构型Ⅰ相比,构型Ⅱ、Ⅲ和Ⅳ的键长均表现出不同程度的变化。其中,N(1)—H(2)键变化最大,构型Ⅱ、Ⅲ和Ⅳ的N(1)—H(2)键分别增长了0.0623,0.0668和0.1834 nm。这是由于NH3OH+中N(1)—H(2)键不稳定,在氢键作用的影响下,N(1)—H(2)键发生断裂生成H+,H+与NO3-在电荷间静电作用下相互吸引,导致在构型形成过程中出现了H原子的转移,从而引起N(1)—H(2)键长的增加。另外,键长N(1)—O(4)、N(7)—O(8)和N(7)—O(9)键长也出现了较大的变化,这些键长的变化均出现在硝酸羟胺构型形成的过程中,表明氢键作用是导致几何参数变化的主要原因。
伴随着键长的变化,硝酸羟胺的键角和二面角也发生了相应的改变。与构型Ⅰ相比,构型Ⅱ、Ⅲ和Ⅳ的键角N(1)—O(4)—H(5)和O(4)—N(1)—H(6)出现了相对较大的变化。另外,负离子上原子的二面角全部接近0°或者180°,说明构型形成过程中并没有对负离子的平面构型产生影响。但是,正离子的共面性所受影响较大,其中构型Ⅱ、Ⅲ和Ⅳ的二面角H(3)—N(1)— O(4)—H(5)和H(6)—N(1)—O(4)—H(5)受到较大影响。
3.2 自然键轨道分析(NBO)在B3LYP/6-311++G(d, p)水平下,对硝酸羟胺构型进行自然键轨道(NBO)分析。表 2中列出了电子供体(Donor)轨道i、电子受体(Acceptor)轨道j以及相互作用的稳定化能E。稳定化能E与相互作用的强度成正比,稳定化能越大,i与j的相互作用越强[19-20]。从表 2中可以看出构型Ⅱ中N(1)的孤对电子(1)对H(2)—O(10)的σ反键轨道的稳定化能为194.4 kJ·mol-1,构型Ⅲ中的N(1)孤对电子(1)对H(2)—O(10)的σ反键轨道的稳定化能为160.2 kJ·mol-1,构型Ⅳ中的O(4)孤对电子(2)对H(2)—O(10)的σ反键轨道的稳定化能为87.6 kJ·mol-1。由此可以看出,构型Ⅱ和Ⅲ的氢键作用主要发生在阳离子的N的孤对电子与阴离子的H—O的反键轨道之间,构型Ⅳ的氢键作用主要发生在阳离子的O的孤对电子与阴离子的H—O反键轨道之间。另外,三个构型的稳定化能之和均在90.0 kJ·mol-1以上,氢键作用较强,稳定化能排列顺序为构型Ⅱ>Ⅲ>Ⅳ。
| 表 2 硝酸羟胺的B3LYP/6-311++G(d, p)自然键轨道分析部分计算结果 Tab.2 Parts of calculated results of hydroxylamine nitrate at B3LYP/6-311++G(d, p) level by NBO analysis |
在硝酸羟胺分子构型形成的过程中,NH3OH+中N(1)—H(2)键不稳定,N(1)—H(2)键发生断裂生成H+,H+与NO3-在电荷间静电作用下相互吸引,导致在构型形成过程中出现了H原子的转移。下面以NH3OH+为研究对象,研究电场对H转移的影响。
采用杂化密度泛函理论B3LYP方法在6-311++G(d, p)水平上对NH3OH+在无场和不同场强下去质子化解离反应的势垒、过渡态的优化构型以及解离产物中H(2)原子上的电荷进行计算,并对能量进行零点能校正,以此来研究电场作用下的H转移。在无场下,NH3OH+去质子化解离反应的产物中H(2)原子上的电荷量为1.0000 e,这说明NH3OH+经过一个过渡态TS生成的产物是NH2OH和H+,说明H转移的路线为NH3OH+→NH2OH+H+,H转移过渡态优化构型的几何参数如表 3所示,活化能为63.9 kJ·mol-1,由于产物的能量比反应物低839.2kJ·mol-1,此反应为放热反应,其逆反应的活化能为903.1kJ·mol-1,因此,产物既具有热力学稳定性,又具有动力学稳定性。另外,构型Ⅱ中N(1)的孤对电子(1)对H(2)—O(10)的σ反键轨道的稳定化能为194.4 kJ·mol-1,构型Ⅲ中的N(1)孤对电子(1)对H(2)—O(10)的σ反键轨道的稳定化能为160.2 kJ·mol-1,构型Ⅳ中的O(4)孤对电子(2)对H(2)—O(10)的σ反键轨道的稳定化能为87.6 kJ·mol-1,均大于63.9 kJ·mol-1,所以硝酸羟胺分子构型形成过程中,首先发生H的转移,生成NH2OH和H+,然后H+和NO3-结合生成HNO3。因此,三种构型不再表现为离子对之间的相互作用,而是NH2OH和HNO3之间的相互作用。由文献[21]可知,硝酸羟胺的分解机理为HAN↔NH2OH+HNO3,NH2OH+HNO3→HONO+HNO+H2O。硝酸羟胺在分解过程中,首先生成NH2OH和HNO3,该分解机理进一步验证了本文优化得到的硝酸羟胺构型。
| 表 3 H转移过渡态构型的键长以及H转移活化能垒 Tab.3 The bond length of configurations of transition states and activation barriers of H atom transfer |
从表 3中可以看出,外电场的加载对H转移有着显著的影响,随着电场强度的增强,NH3OH+的解离反应的产物中H(2)原子的电荷量均为1.0000 e,表明NH3OH+在有场下的解离机理与无场下相同,去质子化解离反应的产物均为H+和NH2OH,即电场的作用下同样出现了H转移,且H转移的路线为NH3OH+→ NH2OH+H+。在正电场方向,随着电场的增强,H转移活化能逐渐减小。当电场强度由0.000 a. u.增长到0.005 a. u.时,H转移活化能由63.9 kJ·mol-1减小到49.6 J·mol-1,过渡态的平衡构型R(1, 2)、R(1, 3)、R(4, 5)逐渐减小,R(1, 4)和R(1, 6)逐渐增大。在负电场方向,H转移活化能比无场下高,反应变得困难。这是由于解离掉的H(2)原子在电偶极场的负极,电场增强使得N—H键断裂越来越困难。总的来说,随着正向电场的增强,H转移活化能垒逐渐减小,有利于硝酸羟胺分子构型的形成。
3.4 外电场对氢键的影响由NBO分析可知,构型Ⅱ、Ⅲ和Ⅳ的氢键作用主要来自于N(1)…H(2)—O(10)键、N(1)…H(2)—O(10)键和O(4)…H(2)—O(10)键。因此,重点研究了电场对N(1)…H(2)—O(10)、N(1)…H(2)—O(10)和O(4)…H(2)—O(10)键长的影响,构型Ⅱ、Ⅲ和Ⅳ氢键键长随外电场的变化关系如图 2所示。

|
图 2 构型Ⅱ、Ⅲ、Ⅳ的键长随外电场的变化关系 Fig.2 The bond length of configuration Ⅱ、Ⅲ、Ⅳ variations with external electric fields |
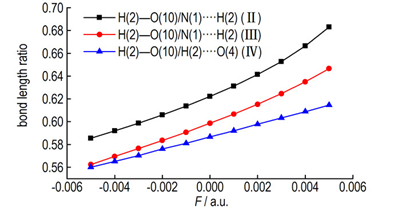
当电场强度由-0.005 a.u.增加到0.005 a.u.时,构型Ⅱ中N(1)…H(2)键长由0.1727 nm减小到0.1555 nm,H(2)—O(10)键长由0.1011增加到0.1062 nm;构型ⅡI中N(1)…H(2)键长由0.1780 nm减小到0.1610 nm,H(2)—O(10)键长由0.1001增加到0.1041 nm;构型Ⅳ中H(2)—O(4)键长由0.1766 nm减小到0.1637 nm,H(2)—O(10)键长由0.0989 nm增加到0.1006 nm,键长的变化与电场强度之间近似呈线性的关系。构型Ⅱ、Ⅲ和Ⅳ中N(1)…H(2)—O(10)键、N(1)…H(2)—O(10)键和O(4)…H(2)—O(10)键的偶极方向相同,所表现出的对电场变化的响应相似。图 3给出了构型Ⅱ中N(1)…H(2)—O(10)键中H(2)—O(10)/N(1)…H(2)的键长比,构型Ⅲ的N(1)…H(2)—O(10)键中H(2)—O(10)/N(1)…H(2)的键长比以及构型Ⅳ的O(4)…H(2)—O(10)键中H(2)—O(10)/H(2)—O(4)的键长比随外电场的变化关系,从图 3中可以看出随着外电场的增加,键长比呈现出增加的趋势,表明键的强度随着电场的增加而增强。另外,键长比的排列顺序为构型Ⅱ>Ⅲ>Ⅳ,表明对电场变化的敏感程度的排列顺序为构型Ⅱ>Ⅲ>Ⅳ。

|
图 3 构型Ⅱ、Ⅲ、Ⅳ的键长比随外电场的变化关系 Fig.3 The bond length ration of configuration Ⅱ、Ⅲ、Ⅳ variations with external electric fields |
结合能随外电场强度的变化如图 4所示,从图 4中可以看出,在无外电场的情况下,构型Ⅱ的结合能绝对值最大,为48.1 kJ·mol-1,分子构型的稳定性最强,构型Ⅱ为硝酸羟胺分子结构的主要存在形式。随着电场的增强,构型Ⅱ、Ⅲ和Ⅳ的结合能表现出单调增大的趋势(数值越负结合能越大)。具体来说,对于构型Ⅱ,当电场强度由-0.005 a.u.增加到0.005 a.u.时,结合能绝对值由34.4 kJ·mol-1增长到66.3 kJ·mol-1;对于构型Ⅲ,当电场强度由-0.005 a.u.增加到0.005 a.u.时,结合能绝对值由24.5 kJ·mol-1增长到56.2 kJ·mol-1;对于构型Ⅳ,当电场强度由-0.005 a.u.增加到0.005 a.u.时,结合能绝对值由13.4 kJ·mol-1增长到51.2 kJ·mol-1。在-0.005~0.005 a.u.电压变化范围内,结合能的变化比较明显。如果将F=0.000 a.u.时的结合能作为参考值,在F=0.005 a.u.时,构型Ⅱ、Ⅲ和Ⅳ的结合能分别增长了18.1 kJ·mol-1、18.1 kJ·mol-1和21.0 kJ·mol-1,即增长了37.715%、47.422%和69.625%。由此可见,电场的变化对结合能产生了显著的影响。另外,结合能绝对值的排列顺序均为Ⅱ>Ⅲ>Ⅳ,由此可见,硝酸羟胺分子稳定性的排列顺序为Ⅱ>Ⅲ>Ⅳ,而且随着电场的增强,硝酸羟胺表现的越来越稳定,形成阴离子或者阳离子变得越来越困难,不利于硝酸羟胺参与化学反应。

|
图 4 结合能随外电场的变化 Fig.4 The binding energy variation with external electric fields |
采用密度泛函B3LYP/6-311++G(d, p)的方法对硝酸羟胺在不同外电场作用下的稳定基态构型进行优化计算,得到了不同外电场作用下的硝酸羟胺分子的最高占据轨道(HOMO)能量EH,最低空轨道(LUMO)能量EL,根据计算公式EG= EL-EH ×27.2 eV求得不同外电场作用下的硝酸羟胺分子的能隙。
最高占据轨道(HOMO)能级反映了失去电子能力的强弱,HOMO能级越高,分子越容易失去电子。最低空轨道LUMO能级在数值上与分子的电子亲和势相当,LUMO能级越低,分子越容易得到电子[22-23]。HOMO与LUMO能量的差值作为能隙EG,EG的大小反映了电子从占据轨道向空轨道发生跃迁的难易程度,在一定程度上代表了分子参与化学反应的能力。
构型Ⅱ、Ⅲ和Ⅳ的能隙随外电场强度变化如图 5所示。随着电场的增强,构型Ⅱ和Ⅳ的能隙不断增大,构型Ⅲ的能隙先增大后减小。具体来说,对于构型Ⅱ和Ⅳ,当电场强度由-0.05 a.u.增加到0.05 a.u.时,构型Ⅱ的能隙由6.0871 eV增大到6.2364 eV,构型Ⅳ的能隙由5.8050 eV增大到6.3199 eV;对于构型Ⅲ,当电场强度由-0.05 a.u.增加到-0.01 a.u.时,构型Ⅲ的能隙由6.4611 eV增大到6.5370 eV,当电场强度由-0.01 a.u.增加到0.05 a.u.时,构型Ⅲ的能隙由6.5370 eV减小到6.4369 eV,在F=-0.01 a.u.时,能隙取得极大值6.5370 eV。由此可见,对于构型Ⅱ和Ⅳ,随着电场的增强,电子从占据轨道向空轨道发生跃迁越来越难,参与化学反应的能力逐渐降低;对于构型Ⅲ,随着电场的增强,电子从占据轨道向空轨道发生跃迁先难后易,参与化学反应的能力先降低后增加。不同电场强度下构型Ⅱ、Ⅲ和Ⅳ的最高占据轨道HOMO和最低未占据轨道LUMO空间分布如图 6所示。在-0.005~0.005 a.u.外电场变化范围内,能隙的变化不是很明显。将F=0.000 a.u.时的能隙为参考值,在F=0.005 a.u.时,构型Ⅱ、Ⅲ和Ⅳ的能隙分别变化了0.0386, 0.0936 eV和0.2053 eV,即变化了0.6228%, 1.4333%和3.3575%。由此可见,电场的变化对能隙的影响有限,即电场对硝酸羟胺化学反应能力的影响较小。

|
图 5 能隙随外电场的变化 Fig.5 The energy gap variation with external electric fields |

|
图 6 前线分子轨道随外电场的变化 Fig.6 Frontier molecular orbital variation with external electric fields |
在-0.005~0.005 a.u.电场强度下,采用Gaussian09软件由B3LYP/6-311++G(d, p)方法研究了硝酸羟胺基推进剂电点火过程中的电场效应对硝酸羟胺的影响,主要得出了以下结论:
(1) 研究结果表明存在三种稳定的硝酸羟胺构型,构型Ⅱ属于CS点群,构型Ⅲ和Ⅳ属于C1点群,与构型Ⅰ相比,构型Ⅱ、Ⅲ和Ⅳ的键长和键角均有不同程度的变化。
(2) 根据NBO分析,构型Ⅱ和Ⅲ的氢键作用主要产生于阳离子的N的孤对电子与阴离子的H—O的反键轨道之间,构型Ⅳ的氢键作用主要产生于阳离子的O的孤对电子与阴离子的H—O反键轨道之间。氢键作用比较强,且稳定化能排列顺序为构型Ⅱ>Ⅲ>Ⅳ。
(3) 沿着正电场方向加载电场,H转移活化能垒逐渐减小,解离反应变得越来越容易,有利于硝酸羟胺分子构型的形成。
(4) 随着电场的增强,硝酸羟胺氢键作用键长缩短,键的强度增强,结合能增加,硝酸羟胺的稳定性增强。但是,电场的变化对能隙的影响有限,即电场对硝酸羟胺化学反应能力的影响较小。
总之,在所研究电场范围内,硝酸羟胺氢键的强度提高,稳定性增强,点火过程中的电场效应对硝酸羟胺的极化或者电离产生负面影响,阻碍了阴离子或者阳离子的形成,对硝酸羟胺发生化学反应产生消极影响,不利于推进剂的点火。
| [1] |
曲艳斌, 肖忠良. 硝酸羟胺水凝胶性能研究[J].
含能材料, 2004, 12(3): 168-170. QU Yan-bin, XIAO Zhong-liang. Study on property of HAN hydrogel[J]. Chinese Journal of Energetic Materials(Hanneng Cailliao), 2004, 12(3): 168-170. |
| [2] |
LIU Li-jun, WEI Chun-yang, GUO Yu-yan, et al. Hydroxylamine nitrate self-catalytic kinetics study with adiabatic calorimetry[J].
Journal of Hazardous Materials, 2009, 162: 1217-1222. DOI:10.1016/j.jhazmat.2008.06.009 |
| [3] |
Barney G S, Duval P B. Model for predicting hydroxylamine nitrate stability in plutonium process solutions[J].
Journal of Loss Prevention in the Process Industries, 2011, 24: 76-84. DOI:10.1016/j.jlp.2010.10.004 |
| [4] |
Ulas A, Boysan E. Numerical analysis of regenerative cooling in liquid propellant rocket engines[J].
Aerospace Science and Technology, 2013, 24(1): 187-197. DOI:10.1016/j.ast.2011.11.006 |
| [5] |
Fu Juan, Chen Xiao-qian, Huang Yi-yong. Validation of a compression mass gauge using groud tests for liquid propellant mass measurements[J].
Advances in Space Research, 2014, 53(9): 1359-1369. DOI:10.1016/j.asr.2014.02.015 |
| [6] |
Dheeraj A, Basu P, Tharakan T J. Prediction of gas-core vortices during draining of liquid propellants from tanks[J].
Aerospace Science and Technology, 2014, 32(1): 60-65. DOI:10.1016/j.ast.2013.12.001 |
| [7] |
Koh K S, Tengku F J C, Chik W K. Role of electrodes in ambient electrolytic decomposition of hydroxylammonium natrate (HAN) solutions[J].
Propulsion and Power Research, 2013, 2(3): 194-200. DOI:10.1016/j.jppr.2013.07.002 |
| [8] |
Amrousse R, Katsumi T, Itouyama N, et al. New HAN-based mixtures for reaction control system and low toxic spacecraft propulsion subsystem: Thermal decomposition and possible thruster applications[J].
Combustion and Flame, 2015, 162(6): 2686-2692. DOI:10.1016/j.combustflame.2015.03.026 |
| [9] |
Keshavarz M H, Zohari N, Seyedsadjadi S A. Relationship between electric spark sensitivity and activation energy of the thermal decomposition of nitramines for safety measures in industrial processes[J].
Journal of Loss Prevention in the Process Industries, 2013, 26(6): 1452-1456. DOI:10.1016/j.jlp.2013.09.012 |
| [10] |
Keshavarz M H. Theoretical prediction of electric spark sensitivity of nitroaromatic energetic compounds based on molecular structure[J].
Journal of Hazardous Materials, 2008, 153: 201-206. DOI:10.1016/j.jhazmat.2007.08.036 |
| [11] |
Lemi T. Contemplation on spark sensitivity of certain nitramine type explosives[J].
Journal of Hazardous Materials, 2009, 169: 454-459. DOI:10.1016/j.jhazmat.2009.03.117 |
| [12] |
王桂香, 肖鹤鸣, 居学海, 等. 含能材料的密度、爆速、爆压和静电感度的理论研究[J].
化学学报, 2007, 65(6): 517-524. WANG Gui-xiang, XIAO He-ming, JU Xue-hai, et al. Theoretical studies on densities, detonation velocities and pressures and electric spark sensitives of energetic materials[J]. Acta Chimica Sinica, 2007, 65(6): 517-524. |
| [13] |
唐翠明, 陈晓旭, 王君, 等. 几种硝铵类炸药在外电场作用下分子的静电火花感度和它们电子性质的关系[J].
四川大学学报, 2013, 50(2): 321-325. TANG Cui-ming, CHEN Xiao-xu, WANG Jun, et al. Relationship between electric spark sensitivity of some nitramines and their electronic properties in external electric field[J]. Journal of Sichuan University, 2013, 50(2): 321-325. |
| [14] |
智春艳, 韩娟, 邱文旭, 等. 硝胺的静电火花感度与分子的电子性质的关系[J].
西安工业大学学报, 2014, 34(9): 698-702. |
| [15] |
Ghazi H E, Jorio A. Excited-states of hydrogenic-like impurities in InGaN-GaN spherical QD: Electric field effect[J].
Physica B, 2013, 430: 81-83. DOI:10.1016/j.physb.2013.08.029 |
| [16] |
Alavi S, Thompson D L. Effects of alkyl-group substitution on the proton-transfer barriers in ammonium and hydroxylammonium nitrate salts[J].
J Phys Chem A, 2004, 108: 8801-8809. DOI:10.1021/jp040189r |
| [17] |
Alavi S, Thompson D L. Hydrogen bonding and proton transfer in small hydroxylammonium nitrate Clusters: A theoretical study[J].
Journal of Chemical Physics, 2003, 119(8): 4274-4282. DOI:10.1063/1.1593011 |
| [18] |
何春芳, 叶近婷, 高阳. 三聚磷酸钠与柠檬酸钠钙螯合机理和螯合能力的对比分析[J].
分子科学学报, 2015, 31(3): 198-202. HE Chun-fang, YE Jin-ting, GAO Yang. Comparative analysis of calcium chelation mechanism and chelating ability about sodium tripolyphosphate and citric acid sodium[J]. Journal of Molecular Science, 2015, 31(3): 198-202. |
| [19] |
方国勇, 徐丽娜, 肖鹤鸣, 等. 3-硝基-1, 2, 4-三唑-5-酮与NH3及H2O分子间相互作用的理论研究[J].
化学学报, 2005, 63(12): 1055-1061. FANG Guo-yong, XU Li-na, XIAO He-ming, et al. Theoretical study on intermolecular interactions of 3-nitro-1, 2, 4-triazol-5-one with NH3 and H2O[J]. Acta Chimica Sinica, 2005, 63(12): 1055-1061. DOI:10.3321/j.issn:0567-7351.2005.12.004 |
| [20] |
徐丽娜, 肖鹤鸣, 方国勇, 等. NTO二聚体分子间相互作用的理论研究[J].
化学学报, 2005, 63(12): 1062-1068. XU Li-na, XIAO He-ming, FANG Guo-yong, et al. Theoretical study on intermolecular interactions of 3-nitro-1, 2, 4-triazol-5-one Dimers[J]. Acta Chimica Sinica, 2005, 63(12): 1062-1068. DOI:10.3321/j.issn:0567-7351.2005.12.005 |
| [21] |
Lee H S, Litzinger T A. Chemical kinetic study of HAN decomposition[J].
Combusition and Flame, 2003, 135: 151-169. DOI:10.1016/S0010-2180(03)00157-3 |
| [22] |
Sakai T, Matsumoto M, Okunishi K, et al. Energy gap of spin nanotube[J].
Physica E: Low-Dimensional Systems and Nanostructures, 2005, 29(3-4): 633-636. DOI:10.1016/j.physe.2005.06.044 |
| [23] |
Horwitz L P, Engelberg E Z. Energy gaps in a spacetime crystal[J].
Physics Letters, 2009, 374(1): 40-43. DOI:10.1016/j.physleta.2009.10.034 |
Three fully optimized geometries of hydroxylamin nitrate ion pair have been obtained with density functional theory method at the B3LYP/6-311++G(d, p) level without the electric field. Natural bond orbital analysis was performed to reveal the origin of the interaction. Bond length, binding energy an level distribution under different electric fields ranging from -0.005 a.u.-0.005 a.u. were calculated.