




含能材料广泛应用于军事和民用领域, 开发研究新型含能材料, 特别是具有高密度、高生成焓、高爆速和爆压且热稳定性好的含能化合物对于含能材料的发展具有重要意义[1-4]。近年来, 五元氮杂环类如呋咱、吡唑、咪唑、三唑和四唑类含能化合物因其具有高的生成焓、良好的氧平衡、富含氮元素等特点受到广泛关注, 大量的工作也表明这类化合物也具有较好的爆轰性能[5-11]。1-甲基-2, 4, 5-三硝基咪唑 (MTNI) 熔点为82 ℃, 被认为是能够替代TNT的下一代熔铸炸药载体炸药[12-13]。5, 5′-联四唑-1, 1′-二氧二羟铵 (TKX-50), 具有与RDX相当的爆轰性能 (理论爆速9.698 km·s-1, 理论爆压42.4 GPa), 但只有与TNT相当的撞击感度 (20 J), 是一种极具潜在应用价值的高能钝感炸药[14-15]。3, 4-二硝基呋咱基氧化呋咱 (DNTF) 不仅具有1.937 g·cm-3的晶体密度, 能量水平与HMX相当, 同时还具有良好的热稳定性[16]。在众多五元氮杂环的研究中, 关于吡咯类化合物的合成的也有相关报道。1966年~1971年Morgan等[17-19]在不同的溶剂体系和宽泛的温度下合成了2-硝基吡咯和3-硝基吡咯, 其熔点分别为65 ℃和101 ℃。1979年, Doddi等[20]先后合成了1-甲基2, 3, 5-三硝基吡咯、1-甲基2, 3, 4-三硝基吡咯和1-甲基2, 3, 4, 5-四硝基吡咯并预言这三种化合物为潜在的含能化合物, 实验测得其熔点分别为111~112 ℃和101~102 ℃。1992年, 2, 3, 4, 5-四硝基吡咯的合成及其X射线单晶衍射数据分别见之报道, 但未有其相关物性报道[21-22]。2015年, Rashid等[23]运用密度泛函理论 (DFT) 预测了2, 3, 4, 5-四硝基吡咯的生成焓。但是关于多硝基吡咯及其衍生物的报道中, 鲜有关于其结构、密度、生成焓、爆轰性能和热安全性的报道。
本论文设计了2, 3, 4, 5-四硝基吡咯 (TNP)、1-甲基-2, 3, 4, 5-四硝基吡咯 (MTNP)、1-氨基-2, 3, 4, 5-四硝基吡咯 (ATNP) 和五硝基吡咯 (PNP) 四种多硝基吡咯化合物, 用密度泛函理论在B3LYP/6-31G*水平对其进行了几何结构全优化, 频率分析无虚频, 得到能量极小值点, 并计算了模型化合物密度。在B3LYP/6-311++G**水平上计算了模型化合物的生成焓、爆轰性能并与已报到的结果进行了比较。通过键离解能的计算和自然键轨道 (NBO) 计算分析模型化合物引发键的强度从而评估了模型化合物的感度, 以期为开发新型含能材料提供了一定的理论参考价值。
2 理论与计算方法使用Gaussian 09软件包[24], 运用密度泛函理论 (DFT) 对TNP、MTNP、ATNP和PNP的结构进行了系统研究, 在B3LYP/6-31G*水平进行了几何结构的全优化。
作为衡量含能化合物能量水平的一个重要特征指标, 密度的大小直接影响了含能化合物的爆速和爆压。根据Kamlet方程[25], 爆压与密度呈正比, 爆速与密度的平方呈正比, 因此在设计新型含能化合物时尤其注重提高化合物的密度水平。本研究在优化结构的基础上, 对模型化合物的密度进行了预测, 在B3LYP/6-31G*水平上对上述分子的体积进行了计算, 进而得到分子理论密度。
生成焓是描述高能量密度材料热力学性质的一个重要参数[26]。由于能量衡算对基组精确度要求较高, 因此本研究选择在B3LYP/6-311++G**水平上通过设计等键反应得到模型化合物的生成焓。按照键离解反应规则, 设计了如式 (1)~式 (4) 的等键反应, 为减小计算误差, 所有计算均保持吡咯环不破坏。
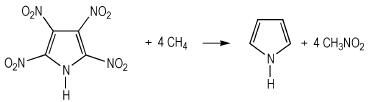
|
(1) |
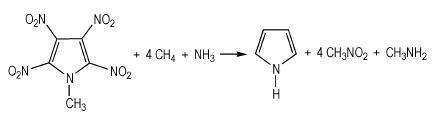
|
(2) |
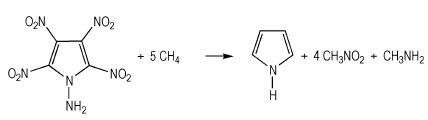
|
(3) |

|
(4) |
298.15 K时, 反应焓变根据式 (5) 计算:
| $ \Delta {H_{{\rm{298}}{\rm{.15 K}}}} = \sum \Delta {H_{{\rm{f, P}}}}-\sum \Delta {H_{{\rm{f, R}}}} $ | (5) |
式中, ΔHf, P与ΔHf, R分别是反应物和产物在298.15 K的生成焓。对于参考物吡咯、CH4、CH3NO2和CH3NH2, 其生成焓的实验值可从文献[27]得到, 因此, 如果能够得到反应的焓变ΔH298.15 K, 则可以计算出模型化合物的生成焓。而ΔH298.15 K可根据如下方程[28-29]计算得到:
| $ \Delta {H_{{\rm{298}}{\rm{.15 K}}}} = \Delta E + \Delta (pV) = \Delta {E_0} + \Delta {\rm{ZPE}} + \Delta {H_{\rm{T}}} + \Delta nRT $ | (6) |
式中, ΔE0为反应物与产物在0 K时总能量之差, a.u.; ΔZPE为反应物与产物的零点能之差, a.u.; ΔHT为反应物与产物从0 K到298.15 K的热力学校正值, a.u., Δ(pV) 则是气体做功, 对于理想气体其等于ΔnRT, 而在等键反应中, Δn=0。
采用Kamlet公式[25]预测爆速和爆压, 如式 (7) 和式 (8):
| $ p = 1.558N{M^{1/2}}{Q^{1/2}}{\rho ^2} $ | (7) |
| $ D = 1.01{\left( {N{M^{1/2}}{Q^{1/2}}} \right)^{1/2}}(1 + 1.30\rho ) $ | (8) |
式中, p为爆压, GPa; D为爆速, m·s-1; N为单位质量炸药爆炸产生的气体产物的摩尔数; M为气体产物的平均摩尔质量; Q为爆热, cal·g-1; ρ为化合物密度, g·cm-3。
为衡量含能化合物的感度, 通常可计算化合物中引发键的强度。而键强可通过键离解能 (BDE) 这一指标衡量。键离解能是断裂一个化学键所需要的总能量[30], 可用式 (9) 计算:
| $ {\rm{BDE}}\left( {A-B} \right) = E\left( {A\cdot} \right) + E\left( {B\cdot} \right)-E(A-B) $ | (9) |
经零点能校正的键离解能[31]为:
| $ {\rm{BDE}}{\left( {A-B} \right)_{{\rm{ZPE}}}} = BDE\left( {A-B} \right) + \Delta {\rm{ZPE}} $ | (10) |
通过自然键轨道 (NBO) 分析, 可得到模型化合物的电子结构特征, 本研究用Gaussian 09 NBO 3.1程序包进行了计算。
3 结果与讨论 3.1 分子结构图 1为优化后TNP、MTNP、ATNP以及PNP的结构示意图, 所有优化计算在B3LYP/6-31G*水平下进行。部分几何参数如表 1所示。TNP中, O (7) 和O (16) 原子与吡咯环基本在同一个分子平面, O (8) 和O (17) 原子向分子平面外旋转了约14°, 这是由于来自邻位硝基 (C (3)—NO2和C (2)—NO2) 的位阻效应, 而这种旋转有利于减小邻位硝基的位阻效应从而使化合物更稳定。同样的原因, O (11) 和O (13) 原子分别向分子平面外旋转了约37°和34°。C (1)—N (5) 键和C (4)—N (5) 键键长均为1.375 Å, 比通常的C—N单键 (1.480 Å) 短0.105 Å, 说明吡咯环内具有一定的共轭效应。O (16) 和O (7) 原子与H (18) 原子距离为2.407 Å, 小于O原子与H原子的范德瓦尔兹半径之和; 同时O (16)—N (5) 和O (7)—N (6) 原子间距离为2.633 Å。这表明, 在TNP分子内存在一个较弱的N—H…O分子内氢键[32-33]。这种分子内氢键的存在有助于提高分子热稳定性。C (1)—NO2和C (4)—NO2键长均为1.443 Å, C (2)—NO2与C (3)—NO2键长均为1.460 Å。可见TNP的分子结构具有一定的对称性。

|
图 1 优化后模型化合物的几何结构 Fig.1 The optimized structures of model compounds |
| 表 1 模型化合物的部分几何参数 Tab.1 The selected parameters of model compounds |
MTNP中, O (16) 原子向分子平面旋转了15°使其更靠近H (20) 原子, 而O (16)—H (20) 原子间距离为2.252 Å, 可见存在一个弱的C—H…O分子内氢键[34]。O (7)—C (18) 原子距离为2.744 Å, 小于范德瓦尔兹半径之和, 表明是一个典型C…O相互作用[35]。ATNP分子中的N (18) 原子向分子平面偏离11°, 而N (18)—N (15) 键长为1.399 Å, 短于通常的N—N单键 (1.45 Å), 表明N (18)—N (5) 相对稳定。O (15)—H (19) 原子间距离为1.906 Å, 明显小于范德瓦尔兹半径之和, 证明是一个强N—H…O分子内氢键。
PNP分子中, N (5)—NO2基团向分子平面外旋转了约83°, 同时其相邻的两个硝基 (C (1)—NO2和C (4)—NO2) 分别向分子平面外旋转了约15°。这种旋转同样能够减小邻位硝基的位阻以使分子更稳定。然而, N (18)—N (5) 键长为1.671 Å, 明显较通常的N—N单键 (1.450 Å) 长, 表明该键相对不稳定。
3.2 生成焓表 2列出了计算得到的电子能量E0(B3LYP/6-311++G**), 零点能ZPE (B3LYP/6-311++G**), 和热力学校正HT(B3LYP/6-31G*)。
| 表 2 化合物E0, ZPE和HOF的计算值与实验值 Tab.2 Calculated E0, ZPE, HOF and experimental gas phase heats of formation for compounds |
TNP、MTNP、ATNP和PNP生成焓的计算值分别为175.6, 193.3, 303.9 kJ·mol-1和341.7 kJ·mol-1。与之前的工作[34]相比, TNP生成焓误差小于2 kJ·mol-1, 表明本研究中计算HOF使用的方法能够得到较为准确、可靠的结果。对于所设计的化合物, HOF与N (1) 取代基的性质相关, 并且HOF随着分子氮含量的增加而升高, 这与通常认为氮含量的提高能够增加化合物的生成焓的认识是一致的。本研究计算得到的MTNP和ATNP的HOF是第一次报道。
3.3 密度和爆轰性能模型化合物以及RDX和HMX的密度和主要爆轰性能参数见表 3。由表 3可知, PNP密度相对最大为2.04 g·cm-3, MTNP密度最小为1.88 g·cm-3。可见硝基的存在能提高分子密度, 甲基的存在会减小分子密度。这种现象在其他氮杂环含能化合物中也有报道[37-38]。
| 表 3 模型化合物及RDX和HMX的性能参数 Tab.3 The properties for model compounds, RDX and HMX |
值得注意的是, 本研究中MTNP的计算值为1.88 g·cm-3, 与文献[22]报道的实测密度 (1.83 g·cm-3) 误差为2.3%。可见本研究计算密度所采用的方法可得到与实际相近的密度值。
由表 3还可知, PNP是四种化合物中唯一具有正氧平衡的含能化合物, 这使得PNP具有高爆压和低爆热的特点, 这对于含能化合物在火炮等武器弹药中的应用极为重要。结合爆速和爆压, TNP则是具有最好爆轰性能的化合物, 而MTNP由于甲基这一非含能基团的存在, 降低了分子密度, 使其能量水平也较低。可见, MTNP能量性能稍差于RDX, 其他三种化合物均高于RDX, 与HMX相当。可见本研究设计的四种化合均为含能化合物, 并且验证了文献[22]的预测: 1-甲基2, 3, 4, 5-四硝基吡咯为潜在含能化合物。
3.4 安全性在B3LYP/6-31G*水平计算得到的四种化合物的硝基电荷和前线道能量及其能级差ΔELUMO-HOMO。由表 4可知, TNP具有最大带隙 (0.16274 a.u.), ATNP则最小 (0.14570 a.u.), 表明TNP较ATNP更稳定。这里的稳定性指的是电子转移或电子激发的化学或光化学稳定性。根据ΔELUMO-HOMO其可能的稳定性为: MTNP>TNP>PNP>ATNP。
| 表 4 模型化合物的硝基电荷及前沿轨道能量 Tab.4 The total nitro charge and frontier orbital energies and their gap of the model compounds |
对于硝基化合物, C—NO2和N—NO2通常是分子中最不稳定的键, 因而分解往往从C—NO2或N—NO2键开始。表 4和表 5显示了各硝基原子的Mulliken电荷和硝基总电荷。在PNP中, N (5)—NO2的总电荷数为正值, 这与通常认为硝基为吸电子基团且带负电荷是不一致的, 可见PNP中N (5)—NO2是分子中最不稳定的 (硝基电荷法[40]认为, 含能化合物所带硝基电荷越负, 硝基越稳定)。因此这四种化合物的稳定性可能为: MTNP>TNP>ATNP>PNP。
| 表 5 部分原子的Mulliken电荷 Tab.5 Mulliken charges on selected atoms of model molecules |
揭示共价键稳定性好坏最根本的办法是计算共价键的键离解能, 即断裂该键所需要的能量大小。键离解能越大说明断裂该键需要更多的能量, 即更不容易断裂, 说明该键越稳定。表 6列出了化合物经NBO分析计算得到的Wiberg键级和BDEs。从表 6可以看出, 除PNP外, TNP、MTNP和ATNP的BDEs均大于120 kJ·mol-1, 满足品优含能材料对其热稳定性的要求 (含能化合物基本的热稳定性要求为BDE大于80 kJ·mol-1, 优含能化合物要求BDE大于120 kJ·mol-1[39])。而PNP中N (5)—NO2键离解能仅60.8 kJ·mol-1, 可见该键是极不稳定的; 并且PNP中N—NO2键级比C—NO2的BDE小, 这与通常认为的N—NO2较C—NO2更不稳定是一致的。根据BDE的大小, 化合物热稳定性可能的顺序为: TNP>ATNP>MTNP>PNP。该结果与模型化合物的分子结构具有一定的相关性。正如前文分析, 在TNP和ATNP分子中存在弱的分子内相互作用, 使得TNP和ATNP分子具有较高的热稳定性。
| 表 6 模型化合物的键级和键离解能 Tab.6 Wiberg bond orders (BO) and BEDs of model compounds |
(1) 在TNP与ATNP存在N—H…O分子内氢键, 并且前者较后者强。TNP分子内同时还存在一种较弱的C…O相互作用, 这些弱的分子内相互作用的存在使得TNP与ATNP具有较好的热稳定性。
(2) 计算得到TNP、MTNP、ATNP和PNP化合物的密度分别为1.96, 1.88, 1.93 g·cm-3和2.04 g·cm-3, 这与通常认为硝基的引入能够提高含能化合物密度是一致的。
(3) 计算得到TNP、MTNP、ATNP和PNP化合物的生成焓分别为175.6, 193.3, 303.9 kJ mol-1和341.7 kJ mol-1。
(4) 计算得到TNP、MTNP、ATNP和PNP化合物的爆速分别为: 9.14, 8.66, 9.01 km·s-1和9.01 km·s-1; 爆压分别为38.95, 34.10, 37.54 GPa和38.73 GPa, 可见MTNP具有与RDX相当的爆轰性能, TNP和ATNP具有与HMX相当的爆轰性能。
(5) 综合爆轰性能和热稳定性分析发现, MTNP具有较好的热稳定性, 满足品优含能材料对热稳定性的要求, 是潜在的含能材料; PNP由于分子内存在5个硝基, 具有较好的爆轰性能但不能达到对含能材料基本热稳定性的要求, 感度差。
| [1] | Agrawal J P, Hodgson R D. Organicchemistry of explosives[M]. England: John Wiley & Sons, 2007: 1-10. |
| [2] | Agrawal J P. High energy materials: propellants explosives and pyrotechnics[M]. England: John Wiley & Sons, 2010: 1-3. |
| [3] | Klapötke T M. High energy density materials[M]. Berlin: Springer Verlag, 2007: 2-4. |
| [4] |
李云路, 薛梅, 王建龙, 等. 多硝基呋咱类含能化合物的合成研究进展[J].
有机化学, 2016, 36(7): 1528-1538. LI Yun-lu, XUE Mei, WANG Jian-long, et al. Advances in the synthesis of poly-nitro furazans[J]. Chinese Journal of Organic Chemistry, 2016, 36(7): 1528-1538. |
| [5] | GaoH X, Shreeve J M. Azole-based energetic salts[J]. Chemical Reviews, 2011, 111(11): 7377 DOI:10.1021/cr200039c |
| [6] | DeHope A, Pagoria P F, Parrish D. New polynitro alkylamino furazans[R]. LLNL-CONF-624954: 2013. |
| [7] | Badgujar D M, Talawar M B, Asthana S N, et al. Advances in science and technology of modern energetic materials: An overview[J]. Journal of Hazardous Materials, 2008, 151(2-3): 289-305. DOI:10.1016/j.jhazmat.2007.10.039 |
| [8] | Talawar M B, Sivabalan R, Mukundan T, et al. Environmentally compatible next generation green energetic materials (GEMs)[J]. Journal of Hazardous Materials, 2009, 161(2-3): 589-607. DOI:10.1016/j.jhazmat.2008.04.011 |
| [9] | Schmitt D, Eyerer P, Elsner P. Insensitive high-performance energetic materials: applied research for optimized products[J]. Propellants, Explosives, Pyrotechnics, 1997, 22(3): 69-87. |
| [10] | LI Bao-hui, SHI Wen-jin, REN Fu-de, et al. A B3LYP and MP2 theoretical investigation on the cooperativity effect; between the X-H center dot center dot center dot H-M (X=F, Cl, Br; M=Li, Na, K) dihydrogen-bonding and H-M center dot center dot center dot; pi interactions involving C6H6[J]. Computational & Theoretical Chemistry, 2013, 1020(2): 81-90. |
| [11] | Fried L E, Ruggiero A J. Energytransfer rates in primary, secondary, and insensitive explosives[J]. Journal of Physical Chemistry, 1994, 98(39): 9786-9791. DOI:10.1021/j100090a012 |
| [12] |
冯露露, 曹端林, 王小军, 等. 1-甲基-2, 4, 5-三硝基咪唑的晶体形貌预测[J].
含能材料, 2015, 23(5): 443-449. FENG Lu-lu, CAO Duan-lin, WANG Xiao-jun, et al. Prediction of crystal morphology of MTNI[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2015, 23(5): 443-449. DOI:10.11943/j.issn.1006-9941.2015.05.008 |
| [13] | Cho J R, Cho S G, Kim K J.et al. A candidate of new insensitive high explosive; MTNI[C]//Insensitive Materials & Energetic Materials Technology Symposium, Enschede, 2000: 393-400. |
| [14] | Fischer N, Fischer D, Klapötke T M. Pushing the limits of energetic materilas: the synthesis and characterization of dihydroxylammonium 5, 5′-bistetrazole-1, 1-diolate[J]. Journal of Material Chemistry, 2012, 22(38): 20418-20422. DOI:10.1039/c2jm33646d |
| [15] |
米向超. 新型高能炸药TKX-50的结晶研究[D]. 太原: 中北大学, 2015.
MI Xiang-chao. Research on the crystallization of the new high explosive TKX-50[D]. Taiyuan: North University of China, 2015. |
| [16] |
胡焕性, 张志忠, 赵凤起, 等. 高能量密度材料3, 4-二硝基呋咱基氧化呋咱性能及应用研究[J].
兵工学报, 2004, 25(2): 155-158. HU Huan-xing, ZhANG Zhi-zhong, ZhAO Feng-qi, et al. A study on the properties and application of high energetic density material DNTF[J]. Acta Armamentarii, 2004, 25(2): 155-158. |
| [17] | Khan M K A, Morgan K J, Morrey D P. Carbonyl derivatives of heterocyclic compounds-Ⅲ[J]. Tetrahedron, 1966, 22(7): 2095-2105. DOI:10.1016/S0040-4020(01)82129-3 |
| [18] | Cooksey A R, Morgan K J, Morrey D P. Nitropyrroles-Ⅱ[J]. Tetrahedron, 1970, 26(21): 5101-5111. DOI:10.1016/S0040-4020(01)93163-1 |
| [19] | Morgan K J, Morrey D P. Nitropyrroles-Ⅲ[J]. Tetrahedron, 1971, 27(1): 245-253. DOI:10.1016/S0040-4020(01)92415-9 |
| [20] | Doddi G, Mencarelli P, Razzini A, et al. Cheminform abstract: nitration of 1-r-pyrrole: formation of polynitro-1-r-pyrroles and orienting effects in the reactions of 3-nitro-1-r-pyrroles[J]. ChemInform, 1979, 10(43): 2199-2924. |
| [21] | Jerald C H, Edward W W. Synthesis of 2, 3, 4, 5-tetranitropyrrole[J]. Journal of Heterocyclic Chemistry, 1992, 29(27): 1721-1724. |
| [22] | Cromer D T, Coburn M D, Ryan R R, et al. Structure of 1-methyl-2, 3, 4, 5-tetranitropyrrole, a possible high-density explosive[J]. Acta Crystallographica Section C, 1986, 42(10): 1428-1430. |
| [23] | Rashid M A M, Cho S G, Choi T H, et al. Heat of formation predictions of various nitro-substituted zaoles by G4MP2-SFM scheme[J]. Theoretical Chemistry Accounts, 2015, 134(11): 1-11. |
| [24] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian[CP], Inc. Wallingford CT, 2013. |
| [25] | Kamlet K M, Jacob S J. Chemistry of detonation a simple method for calculating detonation properties of C—H—N—O explosives[J]. Journal of Chemical Physics, 1967, 48(1): 23-35. |
| [26] | Wang F, Du H C, Zhang J Y, et al. Comparative theoretical studies of energetic azo s-triazines[J]. Journal of Physical Chemistry A, 2011, 115(42): 11852-11860. DOI:10.1021/jp206756r |
| [27] | Mortimer R G. Physical Chemistry[M]. Oxford: Elsevier. Academic Press, 2008. |
| [28] | Zhou Y, Long X P, Shu Y J. Theoretical studies on the heats of formation, densities, and detonation properties of substituted s-tetrazine compounds[J]. Journal of Molecular Modelling, 2009, 16(16): 1021-1027. |
| [29] | Ju X H. Theoretical study on thermodynamic and detonation properties of polynitrocubanes[J]. Propellants, Explosives, Pyrotechnics, 2009, 34(34): 106-109. |
| [30] | Politzer P, Murray J S. Relationships between dissociation energies and electrostatic potentials of C—NO2 bonds: applications to impact sensitivities[J]. Journal of Molecular Structure, 1996, 376(1-3): 419-424. DOI:10.1016/0022-2860(95)09066-5 |
| [31] | Steward J J P. Optimization of parameters for semiempirical methods Ⅰ[J]. Journal of Computational Chemistry, 1989, 10(2): 209-220. DOI:10.1002/(ISSN)1096-987X |
| [32] | Carpenter J E, Weinhold F. Analysis of the geometry of the hydroxymethyl radical by the "different hybrids for different spins" natural bond orbital procedure[J]. Journal of Molecular Structure, 1988, 169: 41-62. DOI:10.1016/0166-1280(88)80248-3 |
| [33] | Bondi A. Van der Waals volumes and radii[J]. Journal of Physical Chemistry, 1964, 68(3): 441-451. DOI:10.1021/j100785a001 |
| [34] | Sutor D J. The C—H…O Hydrogen Bond in Crystals[J]. Nature, 1962, 195(4836): 68-69. DOI:10.1038/195068a0 |
| [35] | Klapötke T M, Mayer P, Schulz A, et al. 1, 5-diamino-4-methyltetrazolium dinitramide[J]. Journal of the American Chemical Society, 2005, 127(7): 2032-2033. DOI:10.1021/ja042596m |
| [36] | Scott A P, Radom L G. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors[J]. Journal of Physical Chemistry, 1996, 100: 16502-16513. DOI:10.1021/jp960976r |
| [37] | Zhang X L, Gong X D. Theoretical study of the stabilities and detonation performance of 5-nitro-3-trinitromethyl-1H-1, 2, 4-triazole and its derivatives[J]. Journal of Molecular Modelling, 2015, 21(2): 1-11. |
| [38] | Zhang X W, Zhu W H, Xiao H M. Theoretical studies on heats of formation, detonation properties, and bond dissociation energies of monofurazan derivatives[J]. International Journal of Quantum Chemistry, 2010, 110(8): 1549-1558. |
| [39] | Chen Z X, Xiao H M. Quantum chemistry derived criteria for impact sensitivity[J]. Propellants, Explosives, Pyrotechnics, 2014, 39(4): 487-495. DOI:10.1002/prep.201300123 |
| [40] | Zhang C Y, Shu Y J, Huang Y G, et al. Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds[J]. Journal of Physical Chemistry B, 2005, 109(18): 8978-8982. DOI:10.1021/jp0512309 |
The structure, detonation properties and safety performance of tetranitropyrrole and its derivatives were determined using DFT method for the first time.