



2. 应用物理与计算数学研究所高性能数值模拟软件中心, 北京 100088
2. Software Center for High Performance Numerical Simulation, Institute of Applied Physics and Computational Mathematics, Beijing 100088, China
近年来, 寻求新一代高能低感且环境友好的绿色含能材料来替代传统的硝胺类炸药一直是含能材料领域的重要研究方向[1]。多氮高能量密度化合物是近年涌现出的一类最具潜力的化合物, 四唑衍生物就是其中的一种。可以通过氧化四唑化合物的氮提高其密度和稳定性, 并降低感度和获得更好的氧平衡, 以满足含能材料高能低感的需求。1, 1′-二羟基-5, 5′-联四唑二羟胺盐(TKX-50)是2012年由德国慕尼黑大学的Fischer等[2]通过在联四唑环上引入氧原子合成的新型高能量密度化合物, 具有较高的氮含量、正的生成焓和较高的密度, 并且具有较好的热稳定性和低的机械感度。理论计算表明, TKX-50的爆速高于1, 3, 5, 7-四硝基-1, 3, 5, 7-四氮杂环辛烷(HMX)和六硝基六氮杂异伍兹烷(CL-20), 撞击感度远低于RDX、HMX和CL-20, 摩擦感度低于或相当于RDX、HMX和CL-20。此外, TKX-50合成容易, 原料低廉, 且毒性较低, 有望成为RDX的优良取代物, 在混合炸药和推进剂领域具有潜在的应用前景。由于其优越的综合性能, 在国内外引起了研究者的广泛关注[3-16]。尽管其合成[2, 6]、热分解机理[4, 8, 13]、分子结构[2]、相容性[3]、热性能[9, 14]、高压结构及稳定性[11-12, 14-16]和以TKX-50为基的高聚物粘结炸药特性[10]等均有人进行了相关的研究, 但是其高压下的力学性能的研究还未见报道, 高压下的结构特性研究也还非常有限。研究压力对TKX-50电子结构和力学性能的影响, 对深入了解TKX-50的微观结构与性能之间的关系具有重要意义。为此, 本研究运用色散校正密度泛函(DFT-D)方法研究了TKX-50在0~100 GPa压力范围的结构、力学性质和电子特性, 第一次运用第一性原理预测了TKX-50的弹性常数。
2 计算方法晶体结构、弹性常数及电子特性计算采用周期性平面波密度泛函理论结合模守恒赝势方法, 采用广义梯度近似(GGA)中的PBE泛函[17]处理相关势能。为考虑分子间的范德华力相互作用, 应用了PBE+Grimme[18]校正方法。TKX-50晶体在布里渊区的倒异空间格点采用Monkhorst-Pack方案[19], 选择k网格点为3×1×2。基组的截断能设置为830 eV。在保持TKX-50晶体的对称性下采用BFGS算法[20]优化晶胞参数和原子坐标。在对晶体进行结构优化计算的过程中, 保证能量收敛标准为5.0×10-6 eV·atom-1, 原子间相互作用力低于0.01 eV·Å-1, 最大应力低于0.02 GPa, 原子最大位移不超过5.0×10-4 Å。在弹性常数的计算过程中, 每个应变模式设置为4步, 最大应变振幅设为0.003, 能量收敛标准设为5.0×10-6 eV·atom-1, 原子间相互作用力低于0.01 eV·Å-1, 原子最大位移不超过5.0×10-4 Å。计算采用CASTEP模块[21]。Hirshfeld面[22]及2D指纹图[23]采用CrystalExplorer3.1[24]计算得到。
3 结果与讨论 3.1 常压及高压下的晶体结构常压下, TKX-50属于对称群为P21/C的单斜晶系。它的晶体结构如图 1所示。首先在单斜晶系对称性限制下, 优化了TKX-50晶体的所有原子和晶格常数。优化得到的晶格常数为a=5.563 Å, b=11.490 Å, c=6.437 Å, β=96.19°。预测的平衡晶胞体积为409.05 Å3, 与基准实验值之间的差值仅为0.13%。模拟得到的晶胞体积和晶格参数与文献的实验值及计算值列于表 1中。由表 1可知, 本研究的计算结果与实验值吻合得很好。说明本计算方法对TKX-50晶体结构及晶体中分子间的相互作用力模拟有较好的可靠性。TKX-50晶体有很强的三维的氢键网络(如图 2所示), 包括了四种类型(N—H…N, N—H…O, O—H…O, 和O—H…N)的8种氢键, 最强的氢键来自羟胺离子上的氢与四唑环上氧, 氢键键长在零压下仅为1.57 Å。随着压力的增加, 晶体中分子的键长均缩短, 这与实验[11]测得的结果一致。

|
图 1 TKX-50的晶体结构(白球代表氢原子, 红球代表氧原子, 篮球代表氮原子, 灰球代表碳原子) Fig.1 Crystal structure of TKX-50 (The white spheres represent the H atoms, the red spheres represent the O atoms, the blue spheres are the N atoms, and the grey spheres are the C atoms) |
| 表 1 计算得到的常压下TKX-50的晶格参数、晶胞体积并与实验值对比 Tab.1 The lattice parameters and unit cell volume (V) of TKX-50 from calculation and comparison with experimental values |

|
图 2 TKX-50的三维氢键网路结构(白球代表氢原子, 红球代表氧原子, 篮球代表氮原子, 灰球代表碳原子, 青绿色虚线代表氢键) Fig.2 Three dimensional hydrogen bonding network structure of TKX-50(The white spheres represent the H atoms, the red spheres represent the O atoms, the blue spheres are the N atoms, and the grey spheres are the C atoms. The cyan dashed lines represent the hydrogen bonds) |
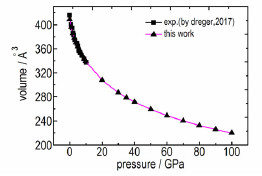
在模拟了常压结构基础上, 研究了0~100 GPa下TKX-50结构的演化。为便于与实验结果比较, 分别在图 3a、图 3c、图 3e和图 3b、图 3d、图 3f中展示了0~11 GPa和0~100 GPa晶格常数和晶胞体积随压力的变化。从图 3a、图 3c、图 3e可以看出, 计算的结果与实验数据[11]吻合很好。在0~100 GPa压力范围内, 晶胞参数(图 3b、图 3d)和晶胞体积(图 3f)的均随压力增加单调变化。此外, 可以发现, 晶轴a轴很难被压缩, b轴最容易压缩。

|
图 3 晶格参数和晶胞体积随压力的变化 Fig.3 Change in lattice parameters and unit cell volume with pressure |
TKX-50中分子间有很强的分子间氢键, 其中典型的4种H…O键和2种H…N键长随压力的变化列于图 4。由图 4可知, 氢键的键长在高压下的压缩率有较大的差异, 有的在低压下压缩率高, 如氢键3在低于10 GPa时, 压缩率远远高于氢键2, 但是在高于20 GPa时, 就很难压缩。部分常压下比较弱的H…N键在高压下迅速被压缩, 如H…N键5。此外, 分析阳离子分子内氢键随压力的变化可知, 在小于10 GPa下, 分子内氢键有所减弱, 但在更高的压力下, 氢键长度又进一步被压缩。

|
图 4 TKX-50中分子间氢键及氢键键长随压力的变化 Fig.4 Change in hydrogen bond and hydrogen-bond length in TKX-50 with pressure |
Hirshfeld面及其指纹图可用于分析晶体结构内部分子间相互作用[25]。Hirshfeld面为通过划分分子相对晶体的电子密度比率为0.5的空间形成的一个连续不重叠的面。TKX-50分子的Hirshfeld面由三维dnorn面生成(图 6所示), 其表面范围为-0.5(红色)~0.5 Å(蓝色)。dnorn面用于鉴别很强的分子间相互作用, dnorn的值用红-蓝-白三种颜色分别代表短于、长于及等于范德华半径长度。在常压和高压下的Hirshfeld面列于图 5。由图 5可知, 随着压力增加, 蓝色区域减小, 红色区域增大, 表明分子间的联系更紧密, 这与晶体在压力下形成更紧密的堆积一致[26]。其详细信息可从相应的2D指纹图(图 6)看出:随着压力增加, 分子间相互作用距离变短, 而且de的最大值也随着压力的增加而减小。

|
图 5 TKX-50在不同压力下的Hirshfeld面 Fig.5 The Hirshfeld surfaces for TKX-50 at different pressures |

|
图 6 TKX-50的在不同压力下的2D指纹图 Fig.6 2D finger print maps of TKX-50 at different pressures |
分子间各种相互作用的比例在0~100 GPa范围随压力的变化见图 7。由图 7可以发现, N…H/H…N氢键对总的Hirshfeld面的贡献最大, 在常压下为总相互作用的42%。随着压力的增加, 氢键相互作用占的比例逐渐降低, 这是由于随着压力增加, 分子间距离减小, 其它弱相互作用有较大的增强空间。

|
图 7 TKX-50各种分子间相互作用随压力的变化 Fig.7 Change in various intermolecular interactions of TKX-50 with pressure |
弹性常数是能量对应变的二阶导数, 它反映材料在不同方向保持弹性的范围。本研究首次用DFT-D方法计算了TKX-50炸药晶体在常压及高压下的13个非零弹性常数, 见表 2。从表 2可知, TKX-50在常压下具有物理稳定性。此外, 弹性常数的三个对角元素C11, C22和C33与三个晶轴方向的分子间相互作用强度相关。三个晶轴方向的弹性常数大小顺序为C33(61.9 GPa)>C11(58.1 GPa)>C22(36.9 GPa), 这归因于c轴方向分子间相互作用最强, 其次是a轴, b轴方向的分子间相互作用最弱。从表 2还可以看出, 随着压力增加, 有10个弹性常数增大。根据单斜晶系的力学稳定性评判标准[27], M6=C66-p>0以及M9=(C33-p)(C55-p)-C352>0, 在0~10 GPa的静水压下, TKX-50在物理上是稳定的。
| 表 2 计算得到的TKX-50晶体在常压及高压下的弹性常数 Tab.2 Elastic constants Cij at atmospheric pressure and high pressure of TKX-50 predicted from calculations |
体积模量B是材料对静水压缩的阻抗, 可通过状态方程的拟合获得。自由能按Eulerian应变(ε)进行展开得到的三阶BM-EOS的能量-体积关系为:
| $ E\left( V \right) = - \frac{9}{{16}}{B_0}\left[{\left( {4-B_0^′} \right)\frac{{V_0^3}}{{{V^{4/3}}}}-\left( {14-3{B^′}} \right)\frac{{V_0^{7/3}}}{{{V^{4/3}}}} + \left( {16 - 3{B^′}} \right)\frac{{V_0^{5/3}}}{{{V^{2/3}}}}} \right] + {E_0} $ |
式中, B0和B′0分别为零压等温体积模量(GPa)及体积模量对压力的一阶偏导, V0为平衡晶胞体积, 根据计算得到的V-E数据点按上述等式采用最小平方差拟合得到B0、B′0和V0。通过在实验晶胞体积左右等间距缩放10个实验晶胞体积进行结构弛豫, 获得的晶胞体积(V, Å3)与能量(E, eV)关系曲线见图 8, 拟合得到的平衡晶胞体积V0为415.1Å3。体模量B0为26.3 GPa, B′0为8.05, 与实验结果[11](B0为21.9 GPa, B′0为8.2)大致吻合。

|
图 8 TKX-50在平衡态附近的E-V曲线 Fig.8 E-V curve near equilibrium state of TKX-50 |
TKX-50在0 GPa、50 GPa和100 GPa下的电子能带结构及其倒易晶格、高对称点和路径见图 9所示。由图 9可知, 随着压力增大, TKX-50的带隙变小, 计算得到常压压力下TKX-50的能带带隙为3.40 eV, 在50 GPa和100 GPa压力下能带带隙分别为3.10 eV和2.75 eV。同时, TKX-50在高压下从直接带隙变为间接带隙。在常压下带隙是从B点到B点的直接带隙, 到50 GPa时已转变为间接带隙, 在50 GPa和100 GPa带隙均为从G点到B点的间接带隙。

|
图 9 TKX-50的能带结构图及对应的倒易晶格 Fig.9 Energy band structure and corresponding reciprocal lattice of TKX-50 |
TKX-50的带隙随着压力增加而减小, 这个变化趋势与其电子密度变化直接相关。很显然, 晶胞体积和分子键长随着压力增大都在减小, 而总的电子数目固定不变, 电荷密度和电子重叠及相互作用均增加, 它们的共同作用提高了化学键力并减小了能带带隙。
爆轰机理研究表明, 能带帯隙的减小可使分子中电子从HOMO到LUMO的转移更容易[30-31]。带隙越小, 电子从价带转移到导带越容易, 进而导致炸药的分解进而产生爆轰。在高压状态下, TKX-50的能带带隙减小, 化学反应活性增大, 从而撞击感度提高。
4 结论(1) 采用基于密度泛函理论加分子间弱相互作用校正方法计算了TKX-50在高压下的结构、力学特性和电子特性, 获得的晶格常数与实验[2]结果有较好的一致性。计算的压缩曲线表明, TKX-50晶体b轴最容易被压缩。首次采用DFT-D计算了TKX-50晶体的常压及高压下弹性常数, 预测得到的TKX-50在常压下的弹性常数大小顺序C33(61.9 GPa)>C11(58.1 GPa)>C22(36.9 GPa), 预测得到的体积模量及其对压力的一阶导数分别为26.3 GPa和8.05, 与文献中[11]实验结果基本符合。
(2) 电子能带结构计算表明, TKX-50的电子特性在常压下带隙是从B点到B点的直接带隙, 到50 GPa时已转变为间接带隙, 在50 GPa和100 GPa带隙均为从G点到B点的间接带隙, 并且带隙随着静水压增加而减小, 预示着TKX-50在高压下撞击感度增大。
| [1] |
Li Y C, Qi C, Li S H, et al. 1, 1′-Azobis-1, 2, 3-triazole:A high-nitrogen compound with stable N8 structure and photochromism[J].
Journal of the American Chemical Society, 2010, 132(35): 12172-12173. DOI:10.1021/ja103525v |
| [2] |
Fischer N, Fischer D, Klapötke T M, et al. Pushing the limits of energetic materials-the synthesis and characterization of dihydroxylammonium 5, 5′-bisterazole-1, 1′-diolate[J].
J Mater Chem, 2012, 22(38): 20418-20422. DOI:10.1039/c2jm33646d |
| [3] |
Huang H F, Shi Y M, Yang J, et al. Compatibility study of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate (TKX-50) with some energetic materials and inert materials[J].
J Energy Mater, 2015, 33(1): 66-72. DOI:10.1080/07370652.2014.889781 |
| [4] |
Yuan B, Yu Z J, Bernstein E R. Initial mechanisms for the decomposition of electronically excited energetic salts: TKX-50 and MAD-X1[J].
J Phys Chem A, 2015, 119(12): 2965-2981. DOI:10.1021/jp510995z |
| [5] |
Niu H, Chen S S, Jin S H, et al. Dissolution properties of dihydroxylammonium 5, 5′-bistetrazole -1, 1′-diolate and disodium5, 5-bistetrazole-1, 1-diolate in water[J].
J Energy Mater, 2016, 34(4): 416-425. DOI:10.1080/07370652.2015.1114048 |
| [6] |
王小军, 苏强, 张晓鹏, 等. 富氮化合物5, 5′-联四唑-1, 1′二氧二羟铵的合成工艺[J].
含能材料, 2015, 23(5): 424-427. WANG Xiao-jun, SU Qiang, ZHANG Xiao-peng, et al. Synthesis of dihydroxylammonium 5, 5′-Bistetrazole -1, 1′-diolate[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2015, 23(5): 424-427. DOI:10.11943/j.issn.1006-9941.2015.05.004 |
| [7] |
Sinditskiia V P, Filatova S A, Kolesova V I, et al. Combustion behavior and physico-chemical properties of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate(TKX-50)[J].
Thermochimica Acta, 2015, 614(1): 85-92. |
| [8] |
An Q, Guang W, William L, et al. Initial steps of thermal decomposition of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate crystals from quantum mechanics[J].
J Phys Chem C, 2014, 118(46): 27175-17181. DOI:10.1021/jp509582x |
| [9] |
Huang H F, Shi Y M, Yang J. Thermal characterization of the promising energetic material TKX-50[J].
Journal of Thermal Analysis and Calorimetry, 2015, 121(2): 705-709. DOI:10.1007/s10973-015-4472-9 |
| [10] |
Niu Hu, Chen Shusen, Shu Qinghai, et al. Preparation, characterization and thermal risk evaluation of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate based polymer bonded explosive[J].
Journal of Hazardous Materials, 2017, 338(complete): 208-217. |
| [11] |
Dreger Zbigniew A, Stash Adam I, Yu Zhi-Gang, et al. High-pressure structural response of an insensitive energetic crystal: dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate (TKX-50)[J].
J Phys Chem C, 2017, 121(10): 5761-5767. DOI:10.1021/acs.jpcc.7b00867 |
| [12] |
Dreger Z A, Tao Y, Averkiev B B, et al. High pressure stability of energetic crystal of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate(TKX-50): Raman Spectroscopy and DFT Calculations[J].
J Phys Chem B, 2015, 119(22): 6836-6847. DOI:10.1021/acs.jpcb.5b02879 |
| [13] |
Lu Zhipeng, Zeng Qun, Xue Xianggui, et al. Does pressure increasing always accelerate the condensed material decay initiated through bimolecular reactions? a case of the thermal decomposition of TKX-50 at high pressures[J].
Phys Chem Chem Phys, 2017, 19(34): 23309-23317. DOI:10.1039/C7CP04015F |
| [14] |
Lu Zhipeng, Xue Xianggui, Meng Liya, et al. Heat-induced solid-solid phase transformation of TKX-50[J].
J Phys Chem C, 2017, 121(15): 8262-8271. DOI:10.1021/acs.jpcc.7b00086 |
| [15] |
Meng Liya, Lu Zhipeng, Ma Yu, et al. Enhanced intermolecular hydrogen bonds rooting for the highly dense packing of energetic hydroxylammonium salts[J].
Cryst Growth Des, 2016, 16(12): 7231-7239. DOI:10.1021/acs.cgd.6b01409 |
| [16] |
Meng Liya, Lu Zhipeng, Wei Xianfeng, et al. Two-sided effects of strong hydrogen bonding on the stability of dihydroxylammonium 5, 5′-bistetrazole-1, 1′-diolate (TKX-50)[J].
Cryst Eng Comm, 2016, 18(13): 2258-2267. DOI:10.1039/C5CE02089A |
| [17] |
Perdew J P, Chevary J A, Vosko S H, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation[J].
Phys Rev B, 1992, 46(11): 6671-6687. DOI:10.1103/PhysRevB.46.6671 |
| [18] |
Grimme S. Accurate description of Van der Waals complexes by density functional theory including empirical corrections[J].
J Comput Chem, 2004, 25(12): 1463-1473. DOI:10.1002/(ISSN)1096-987X |
| [19] |
Monkhorst H J, Pack J D. Special points for brillouin-zone integrations[J].
Phys Rev B, 1976, 13(12): 5188-5192. DOI:10.1103/PhysRevB.13.5188 |
| [20] |
Pfrommer B G, Cote M, Louie S G, et al. Relaxation of crystals with the quasi-Newton method[J].
J Comput Phys, 1997, 131(1): 233-240. DOI:10.1006/jcph.1996.5612 |
| [21] |
Clark S J, Segall M D, Pickard C J, et al. First principles methods using CASTEP[J].
Z Kristallogr, 2005, 220(5-6): 567-570. |
| [22] |
McKinnon J J, Spackman M A, Mitchell A S, et al. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals[J].
Acta Crystallogr Sect. B, 2004, 60(6): 627-668. DOI:10.1107/S0108768104020300 |
| [23] |
Spackman M A, McKinnon J J. Fingerprinting intermolecular interactions in molecular crystals[J].
CrystEngComm, 2002, 4: 378-392. DOI:10.1039/B203191B |
| [24] |
Wolff S K, Grimwood D J, McKinnon J J, et al. CrystalExplorer[CP]. University of Western Australia, 2005.
|
| [25] |
Hirshfeld F L. Bonded-atom fragments for describing molecular charge densities[J].
Theor Chim Acta, 1977, 44(2): 129-138. |
| [26] |
Spackman Mark A, McKinnon Joshua J, Jayatilaka D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals[J].
Cryst Eng Comm, 2008, 10(4): 377-388. |
| [27] |
Liu Q J, Zhang N C, Sun Y Y, et al. Density-functional theory study of the pressure-induced phase transition in hydronitrogen compound N4H4[J].
Phys Lett A, 2014, 378(18-19): 1333-1335. DOI:10.1016/j.physleta.2014.03.024 |
| [28] |
Zhu W, Xiao J, Ji G, et al. First-Principles study of the four polymorphs of crystalline octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine[J].
J Phys Chem B, 2007, 111(44): 12715-12722. DOI:10.1021/jp075056v |
| [29] |
Zhu W, Zhang X, Wei T, et al. DFT studies of pressure effects on structural and vibrational properties of crystalline octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine[J].
Theor Chem Acc, 2009, 124(3-4): 179-186. DOI:10.1007/s00214-009-0596-y |
| [30] |
Luty T, Ordon P, Eckhardt C J. A model for mechanochemical transformations: Applications to molecular hardness, instabilities, and shock initiation of reaction[J].
J Chem Phys, 2002, 117(4): 1775-1785. DOI:10.1063/1.1485968 |
| [31] |
Kuklja M, Stefanovich E, Kunz A. An excitonic mechanism of detonation initiation in explosives[J].
J Chem Phys, 2000, 112(7): 3417-3423. DOI:10.1063/1.480922 |
The structure, mechanical properties, intermolecular interactions and electronic properties of dihydroxylammonium 5,5′-Bistetrazole-1,1′-diolate(TKX-50) under high pressure are thoroughly studied based on the accurate prediction of pressure-volume relationship.