



1, 1-二氨基-2, 2-二硝基乙烯(FOX-7)能量水平与黑索今(RDX)相当,但感度与三氨基三硝基苯(TATB)等钝感炸药相近,是极具应用前景的高能钝感含能材料。FOX-7热解理论的基础研究可深入认识其结构与性能的关系,可对含能材料分子设计、实际应用提供理论指导。近年来密度泛函理论已经成为研究含能材料热解机理的有力工具。
研究FOX-7的感度时,Helen Dorsett[1]在B3LYP/cc-pVDZ水平上计算发现,FOX-7中硝基氧原子与邻近的氨基氢原子之间距离约1.8 Å,两基团之间存在的分子内氢键是导致FOX-7裂解活化能升高的因素之一。Peter Polizer等[2]在研究乙烯硝基/氨基衍生物分子结构稳定性时,发现FOX-7分子中存在的“推-吸电子效应”对其结构稳定具有重要作用。由此可见,分子内基团间的相互作用会导致裂解活化能变化。S.N.Rashkeev等[3]采用第一性原理研究了FOX-7晶相电子结构及裂解,发现FOX-7晶相与单分子有着截然不同的裂解机理,且理想晶型与存在缺陷的晶型裂解机理也不相同。不同状态的FOX-7有不同的分子间相互作用,可见分子间相互作用也会影响其裂解机理。但是,分子间相互作用对FOX-7各裂解通道影响的定量理论研究,至今未见相关报道。
目前, 凝聚相FOX-7的裂解机理大都采用分子动力学(MD)方法研究,MD方法依托牛顿力学构建势函数,无法准确描述与电子运动相关的分子间相互作用,而密度泛函理论可描述多体电子运动,揭示分子间相互作用的本质。本研究采用色散校正密度泛函理论(DFT-D3),用FOX-7的二聚体和三聚体模拟其分子在晶体中的存在状态,以此阐述凝聚相FOX-7的分子间相互作用,并研究分子间相互作用对FOX-7裂解的影响。
2 计算方法DFT-D3理论[4]采用类似分子力场的方法引入色散项来校正长程交换相关势,解决了传统交换相关泛函描述分子间相互作用时的局限。本研究采用ORCA程序[5],所有计算均利用DFT-D3理论的RI-B2PLYP-D3和PW6B95-D3方法,使用def2-TZVP基组和def2-TZVP/J辅助基组; BSSE采用gCP法[6]结合DFT/TZVP水平校正。在得到各团簇稳定构型后,对各团簇裂解通道上的反应物、过渡态和产物进行全参数优化,得到最优几何构型。对所有结构进行频率分析,确认反应物和产物在势能面的极小值点,过渡态在一阶鞍点。涉及过渡态的反应计算内禀反应坐标(IRC),使每个过渡态连接的2个极小点是相应的反应物和产物。团簇形成前后的电子密度差图和Hirshfeld surface指纹图使用Multiwfn程序[7]绘制。
3 结果与讨论 3.1 团簇构型为了尽可能真实地研究FOX-7分子间相互作用,在X-ray衍射法得到的晶体结构[8]中截取FOX-7分子团簇构型,并采用PW6B95-D3方法全参数优化,得到如图 1所示的四种团簇构型。

|
图 1 FOX-7团簇构型(黑色数字代表原子间距离/Å) Fig.1 FOX-7 clusters configurations (Black numbers represent the distance between atoms in Å) |
Ⅰ与Ⅱ是气相FOX-7二聚体的两种构型,其中团簇Ⅰ是平面头尾相接模式,从原子间距离来看,存在分子间相互作用的有H(12)与O(24)及H(13)与O(23),原子间距离分别为2.403 Å和2.093 Å。团簇Ⅱ是平行错位堆积模式,O(23)与H(13)和H(14)之间的距离分别为2.326 Å和3.058 Å,极有可能存在分子间相互作用。
Ⅲ与Ⅳ是FOX-7三聚体的两种构型,其中团簇Ⅲ可看作是团簇Ⅰ与Ⅱ的叠加模式,更全面地展示了FOX-7分子间相互作用。团簇Ⅰ,Ⅱ和Ⅲ中FOX-7分子的存在方式与晶体中极其类似。但是团簇Ⅳ中三个FOX-7分子形成了一种类笼状结构,三个分子头尾相接,从原子距离来看,其间涉及复杂的分子间相互作用。虽然团簇Ⅳ与晶体中分子的排列方式截然不同,但由于其形成了类笼状的三维结构,其构型可能相对稳定,FOX-7热解时晶体结构被破坏后,高温高压的气相状态下极有可能形成此种构型。居学海等[9]研究FOX-7二聚物时也发现了与Ⅰ,Ⅱ类似的结构,并且计算了二聚物形成过程中的吉布斯自由能,发现与团簇Ⅰ类似的结构在常温下可自发形成。由图 1可知,FOX-7分子在团簇中的相互作用方式与晶体结构中基本相同,能基本再现FOX-7晶体中分子的存在状态,以研究分子间相互作用对裂解的影响。
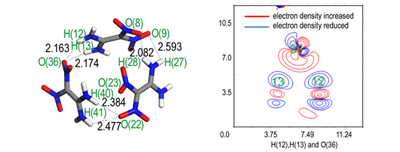
3.2 电子密度变化分析电子密度差图可以反映复合物形成前后复合物所在空间电子密度的变化,可直观展示化学键及分子间相互作用的形成。图 2为FOX-7团簇中分子间相邻处的电子密度差图。

|
图 2 FOX-7团簇中分子间相邻处电子密度差图(绿色数字代表与图 1对应的原子编号,坐标轴为分子空间相对位置/Bohr) Fig.2 Electron density difference between adjacent molecules in FOX-7 clusters (The green number represents the atomic number corresponding to Fig. 1, coordinate axis represents the relative position of the molecular space/Bohr) |
图 2a是团簇Ⅰ中通过H(12)、H(13)和O(23)所在平面的电子密度差图。两个FOX-7分子接近而形成团簇的过程中,O(23)附近电子受到氢核的吸引发生了明显的极化,导致部分电子向H(12)和H(13)偏移,而H(12)和H(13)附近的电子又受到O(23)附近电子的静电排斥而继续向氨基上N原子方向偏移。如此,由图 2a可以看出两个FOX-7分子形成团簇的过程中电子有固定的转移方向。而这一系列的电子转移,造成了H(12)和H(13)与O(23)之间出现了一块电子密度增加的区域,形成了部分的共享电子。从气相分子团簇的角度来看,两个FOX-7分子通过分子间作用形成了分子团簇体系; 从FOX-7层状的晶体结构来看,各层内部是通过定向分子间电子转移而形成的稳定结构。原本分子内的电子向分子间偏移,形成分子间相互作用,从能量守恒角度来看,这必然是以削弱分子内部分键为代价的。
图 2b是团簇Ⅱ中通过H(13)、H(14)和O(23)所在平面的电子密度差图,O(23)的电子向H(13)方向偏移,H(13)与O(23)之间出现了明显的共享电子区域,产生了分子间相互作用。团簇Ⅱ是二聚物的平行错位堆积模式,分子间相互作用力比较简单,电子由O(23)向H(13)偏移。
图 2c~图 2e是团簇Ⅲ中涉及分子间相互作用的各平面的电子密度差图。团簇Ⅲ是Ⅰ与Ⅱ叠加而成,可全面展示晶体状态时FOX-7分子间相互作用。由图 2a分析可知,FOX-7平面头尾相接模式中,电子有确定的转移方向,此处着重分析层间电子偏移。由图 2c、图 2d和图 2e可知,电子分别从O(9)向H(26)偏移,O(22)向H(12)偏移,O(36)向H(28)偏移。结合图 1(Ⅲ)团簇分子构型可知,在团簇形成过程中电子在各层之间相互转移,电子的层间转移并无特定方向。
图 2f~图 2i是团簇Ⅳ中涉及分子间相互作用的各平面的电子密度差图。分析图中电子偏移方向可知,三个FOX-7分子形成类笼状的分子团簇时,电子分别从O(8)向H(28),O(9)向H(27),O(22)向40 H(41)和H(42),O(36)向H(12)和H(13)偏移。从图 1(Ⅳ)的角度看,3个FOX-7分子头尾相接形成了类笼状结构,导致电子整体以顺时针方向偏移,产生闭合环状电子流,形成相对稳定分子团簇体系。
3.3 裂解通道活化能FOX-7最有可能的初始裂解通道是C—NO2键均裂和硝基异构[2],表 1为采用密度泛函PW6B95-D3和RI-B2PLYP-D3理论,计算的四种FOX-7分子团簇中C—NO2键均裂和硝基异构(表 1中以ONO表示)通道的活化能,以此考察分子间相互作用对FOX-7裂解活化能的影响。为充分说明分子间相互作用对裂解的影响,团簇Ⅰ和Ⅱ的两种通道计算O(23)所在硝基,团簇Ⅲ计算O(22)所在硝基,团簇Ⅳ计算O(36)所在硝基。
| 表 1 不同泛函计算所得活化能 Tab.1 Activation energy obtained by different functional calculation |
在RI-B2PLYP-D3和PW6B95-D3两种不同方法下,四种团簇各裂解通道的活化能变化趋势一致,以下讨论以PW6B95-D3方法为例。由表 1可知,与单分子FOX-7相比,四种团簇C—NO2键均裂的活化能均有不同程度的降低。团簇Ⅱ计算的O(23)所在硝基与团簇Ⅲ计算的O(22)所在硝基位置相似,所受到的分子间相互作用也相似。由图 2b的电子密度分析可知,团簇Ⅱ中O(23)的部分电子向H(13)偏移,由此导致束缚在C—NO2键邻域的电子向N—O键偏移,而C—C键所偏移电子又无法有效补偿,必然使C—NO2键弱化。所以,团簇Ⅱ、Ⅲ中C—NO2键均裂活化能分别下降18.4和22.2 kJ·mol-1。团簇Ⅰ与团簇Ⅳ的C—NO2键均裂活化能受分子间相互作用影响较小。由图 2a可知,在团簇Ⅰ中,O(23)发生了电子偏移,与邻近的H(12)和H(13)同时发生了分子间相互作用,一方面C—NO2键受到弱化,另一方面C—NO2键断裂的过程中不可避免的受到了来自邻近FOX-7分子的空间位阻,两种作用相互抵消,使团簇Ⅰ的C—NO2键均裂活化能变化不大。
团簇Ⅰ与团簇Ⅳ硝基异构通道的活化能有所升高,尤其是团簇Ⅳ较单分子升高了39.4 kJ·mol-1(表 1)。团簇Ⅱ硝基异构通道的活化能下降了210.9 kJ·mol-1,团簇Ⅲ变化不明显,较单分子仅降低了5.3 kJ·mol-1。硝基异构反应首先是整个硝基沿C—N轴旋转90°左右[10],由图 2h可知,在团簇Ⅳ中,O(36)所在硝基受到的分子间作用力正好束缚了C—N轴的旋转,从而使团簇Ⅳ硝基异构通道的活化能大幅升高。而由图 2b可知,在团簇Ⅱ中O(23)所在硝基受到的分子间作用力恰好与C—N轴旋转方向一致,且在形成团簇Ⅱ时O(23)所在硝基就已经旋转了一定的角度,这才使团簇Ⅱ硝基异构通道活化能大幅下降。
结合图 2电子密度分析可知,在团簇形成的过程中,各FOX-7分子之间形成了部分分子间共享电子,这部分电子由原来的单分子FOX-7内部转移到了分子之间,形成分子间相互作用的同时也弱化了分子内的部分化学键。但由于团簇中各基团空间位置和角度的不同,受到不同的空间位阻作用,使FOX-7各裂解通道的活化能呈现不同的变化。
4 结论(1) 在RI-B2PLYP-D3和PW6B95-D3方法下,由FOX-7晶体结构得到了四种团簇构型,分子在团簇中的相互作用方式与晶体结构中基本相同,能基本再现晶体中FOX-7分子的存在状态。
(2) FOX-7团簇中分子间相互作用源于电子偏移形成的部分分子间共享电子。
(3) 团簇中分子间相互作用的存在使C—NO2键均裂的活化能较单分子FOX-7有所降低。但由于分子间相互作用力角度的影响,在采用PW6B95-D3方法时,团簇Ⅱ硝基异构通道的活化能较单分子FOX-7下降了210.9 kJ·mol-1,团簇Ⅳ硝基异构通道的活化能较单分子FOX-7升高了39.4 kJ·mol-1。
| [1] |
Helen D. Computational studies of FOX-7, a new insensitive explosive, DSTO-TR-1054[R], 2000.
|
| [2] |
Peter P, Monica C C, Grice M E, et al. Computational investigation of the structures and relative stabilities of amino/nitro derivatives of ethylene[J].
Journal of Molecular Structure:Theochem, 1998, 452: 75-83. DOI:10.1016/S0166-1280(98)00136-5 |
| [3] |
Rashkeev S N, Kuklja M M, Zerilli F J. Electronic excitations and decomposition of 1, 1-diamino-2, 2-dinitroethylene[J].
Applied Physics Letters, 2003, 82(9): 1371-1373. DOI:10.1063/1.1557768 |
| [4] |
Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory[J].
Journal of Computational Chemistry, 2011, 32(7): 1456-1465. DOI:10.1002/jcc.v32.7 |
| [5] |
Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J].
Journal of Computational Chemistry, 2006, 27(15): 1787-1799. DOI:10.1002/(ISSN)1096-987X |
| [6] |
Kruse H, Grimme S. A geometrical correction for the inter-and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems[J].
The Journal of Chemical Physics, 2012, 136: 154101 DOI:10.1063/1.3700154 |
| [7] |
Lu T, Chen F W. Multiwfn: a multifunctional wavefunction analyzer[J].
Journal of Computational Chemistry, 2012, 33: 580-592. DOI:10.1002/jcc.v33.5 |
| [8] |
Evers J, Klapötke T M, Mayer P, et al. α-and β-FOX-7, polymorphs of a high energy density material, studied by X-ray single crystal and powder investigations in the temperature range from 200 to 423K[J].
Inorganic Chemistry, 2006, 45(13): 4996-5007. DOI:10.1021/ic052150m |
| [9] |
Ju X H, Xiao H M, Xia Q Y. A density functional theory investigation of 1, 1-diamino-2, 2-dinitroethylene dimers and crystal[J].
The Journal of Chemical Physics, 2003, 119(19): 10247-10255. DOI:10.1063/1.1618735 |
| [10] |
李小东, 徐哲, 王晶禹, 等. 带电粒子对FOX-7裂解通道影响的密度泛函理论研究[J].
含能材料, 2017, 25(01): 26-31. LI Xiao-dong, XU Zhe, WANG Jing-yu, et al. Density functional study of the effect of charged particles on FOX-7 dissociation channels[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2017, 25(01): 26-31. |
The research of FOX-7 clusters by using dispersion corrected density functional theory was performed to simulate the existing state of FOX-7 molecule in crystal structure. Electron density difference at binding sites in FOX-7 clusters was plotted. The intermolecular interaction of condensed phase FOX-7 and its effects on the decomposition mechanism of FOX-7 were also described.