




2. School of Materials Science and Engineering, Nanjing Institute of Technology, Nanjing 211167, China
2. 南京工程学院材料科学与工程学院,江苏 南京 211167
It is particularly a challenge to obtain a clear picture on the decomposition mechanisms of condensed phase explosives under extreme conditions due to their complex behaviors and danger in experimental measurements The atomistic simulation is an effective way to model the physical and chemical properties of complex solids under extreme conditions at the atomic level as a complement to experimental work. Recently, the application of ab initio molecular dynamics (AIMD) method in studying the decomposition mechanisms and sequences of the explosives at realistic reaction temperatures and pressures has seen tremendous progress[1-4].
1, 3, 5-Trinitro-1, 3, 5-triazocyclohexane (RDX) is one of the most important nitramine explosives widely used for both civilian and military applications. The decomposition of RDX has been investigated by a number of groups using both experimental and theoretical methods[5-11]. These studies, which were conducted under a variety of ambient- and high pressure environments, discuss a number of decomposition mechanisms that RDX may undergo; these include the loss of NO2 groups[11], the cleavage of the C—N bond to create CH2—N2—O2 species, as well as whether the decomposition is a unimolecular or bimolecular process[12]. There is an obvious difference between the two researches, so it is necessary to study the initial and subsequence decomposition mechanisms of RDX under extreme conditions further. In this work, AIMD was used to systematically study the reactive events of the initial decomposition and following reactions of crystalline α-RDX under high temperature (3000 K, which is the flame temperature of α-RDX) coupled with high pressure (34.5 GPa, which is the detonation pressure of α-RDX).
2 Simulations and Computational MethodAll calculations were performed in the framework of DFT[13-14] using norm-conserving pseudopotentials[15] and a plane-wave expansion of the wave functions based on the CASTEP code[16]. The exchange and correlation were treated with the generalized gradient approximation (GGA), using the functional form of Perdew-Burke-Ernzerhof[17] (PBE) method. To correct DFT for neglecting van der Waals (vdW) interaction, Grimme (G06)[18] and Tkatchenko and Scheffler (TS)[19] correction to the PBE functionals (labeled as PBE-G06 and PBE-TS) were used. RDX belongs to the orthorhombic space group P21/n with experimental lattice constants (a=13.18 Å, b=11.57 Å, c=10.71 Å) [20]. One unit cell consists of four molecules (168 atoms). Both geometry and cell volume were optimized using periodic DFT with vdW correction (DFT-D). A plane wave cutoff of 500 eV for molecular dynamics and 750 eV for geometry and cell optimizations was utilized. Constant temperature AIMD simulations were performed using the Car-Parrinello scheme[21] with Nosé-Hoover thermostats[22] for nuclei. Time steps 1 fs and 2 fs were used for the integration of the equations of motion during production and equilibration runs, respectively. The results indicate that 1 fs is much more suitable. The time step of 1 fs is chosen to longer time simulation of 20 ps. The convergence criteria were set to a 1×10 -6 eV energy difference for geometry optimization and a 1×10 -5 eV energy difference for DFT-MD simulation. he structures for the α-RDX were optimized individually before AIMD simulations. The procedure for the DFT-MD simulations was as follows: First, the system was equilibrated at 298.15 K for 10 ps using NVT. Then, AIMD simulations were carried out using NPT at 3000 K coupled with 34.5 GPa based on this equilibrated system. The simulation time was 20 ps for the AIMD simulations.
3 Results and DiscussionHere, the PBE-G06 and PBE-TS functionals to fully relax α-RDX crystals at ambient pressure without any constraint were applied, respectively. Table 1 lists the experimental and relaxed cell parameters of α-RDX crystal. It is found that calculated lattice parameters by the PBE-TS functional are 2.35%, 0.86%, 1.68% and 4.93% over the experimental parameters. The PBE-G06 calculation errors are 0.99%, 0.26%, 0.56%, and 1.72% for lattice constants a, b, and c, respectively. This indicates that the lattice parameters estimated by the PBE-G06 here are much closer to the experimental value than those by the PBE-TS. Therefore, the PBE-G06 functional is much suitable for studying α-RDX.
| Tab.1 Comparison of relaxed lattice parameters of α-RDX with experimental data under ambient pressure conditions |
At 3000 K coupled with 34.5 GPa, the initial decomposition step of α-RDX is the unimolecular C—H bond breaking to form hydrogen radical (Fig. 1). The H radicals then induced the cleavage of two C—N bonds in other α-RDX molecule. The bond lengths of the two C—N bonds increase to 1.857 Å and 1.762 Å at first 3 fs, respectively. This suggests that the H radicals play a catalytic role in promoting subsequent decompositions.

|
Fig.1 Initial decomposition mechanisms of α-RDX crystal at high temperature 3000 K coupled with detonation pressure 34.2 GPa |
To identify the breaking of intermolecular C—H bond, the density of states (DOS) of C and H atoms in RDX was investigated. Fig. 2 displays the DOS of the C and H states before and after the decomposition of α-RDX. It is found that the variation trends for the DOS curves of the C and H states before the decomposition of α-RDX are similar. But they have a few changes after decomposition. Three main strong peaks occur almost at the same energy in the DOS of the C and H states before and after decomposition of α-RDX. Although their DOS peak height is almost the same before its decomposition, their peaks obviously reduce after its decomposition.

|
Fig.2 DOS curves of the C and H states in the C—H bond before and after the initial decomposition of α-RDX |
This may be due to the C—H bond cleavage in the decomposition process. Fig. 3 displays the DOS of the C and N states before and after the decomposition of α-RDX. It is seen that the variation tendency for the DOS of the C and N states before the decomposition of α-RDX is similar. All of the peaks occur at the same energy in the DOS of the C and N atoms states before its decomposition. This indicates that the two atoms are strongly bonded. However, after the decomposition of α-RDX, some main peaks do not occur at the same energy. This clearly validates that the C—N bond is broken.

|
Fig.3 DOS curves of the C and N states in the C—N bond before and after the initial decomposition of α-RDX |
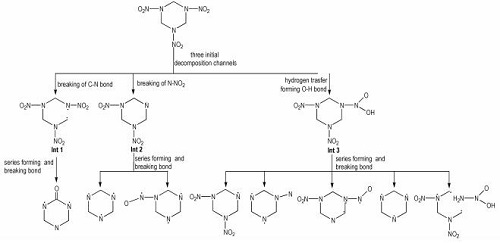
After the initial decomposition, three subsequent decomposition processes take place, as displayed in Fig. 4. Three main interesting decomposition paths include: (1) the C—N bond hemolysis triggers the other C—N bonds of this ring to break; (2) the dissociation of N—NO2 bond releases NO2 gas; (3) the H radical attacks the O atom to release O—H radical by forming O—H bond. However, the decomposition processes are different from reported four different primary decomposition pathways[23-24]. Although our primary decomposition pathways do not include successive HONO elimination[25], there exist the HONO fragments in decomposition products of α-RDX. In our simulation, it is not observed that an oxygen migrates from NO2 to its neighboring C tom, which was reported by Botcher et al[24]. There are a large number of subsequent generated intermediates in the unimolecular decomposition of α-RDX. Among them, Int 1, Int 2, and Int 3 are main intermediates. Compared with Int 2 and Int 3, the incidences of Int 1 are very low. This indicates that the N—NO2 fragments are very active. There are several different heterocyclic compounds observed in the decomposition. These heterocyclic products can transform to form chain fragments. The heterocyclic C3N3 is most likely to occur. This demonstrates that the heterocyclic C3N3 is much more stable than the N—NO2 fragments.

|
Fig.4 Three subsequent decomposition paths after the initiation of α-RDX crystal |
Fig. 5 shows the time evolution of the population of key decomposition products during the cleavage of α-RDX. The RDX molecules delayed quickly at the beginning of the decomposition. All the nitro groups released fast at about 0.05 ps and the number of the nitro groups reaches a maximum of 10. Then the NO2 radicals decayed quickly within about 0.1 ps. At the same time, the number of HONO radicals increased to reach a maximum of 5 at about 0.05 ps. As the HONO is a very active intermediate, it decomposed rapidly to yield HO and NO fragments. The concentration of HONO was consumed practically during the time of about 0.1 ps. After the NO2 and HONO radicals disappeared, nitric oxide accumulated quickly to reach a maximum concentration of 5 molecules within about 0.1 ps. Afterwards they slowly reacted to form N2, N2O, and NOH. Fig. 5 provides a proof for the NO2 and HONO intermediates decomposing to release NO. NO2 can react with H to form NO. At the decomposition end of the NO2 radicals, the concentration of N2O4 reaches the maximum. But they strongly fluctuated due to high oxidization activity. N2O4, a prototypical species of weakly bonded systems, has been the subject of intense theoretical interest[26-28]. NO2 radical is known to exist in the crystal as a dimer N2O4 and forms via addition of two NO2 radicals[29].

|
Fig.5 Time evolution of the number of RDX molecule, H2O, N2, O—H, NO2, CO2, H, NO, HONO, N2O2 and N2O during the whole decomposition stage of RDX crystal |
At about 2 ps, the concentration of nitrous oxide reaches the maximum, but they strongly fluctuates due to high oxidization activity. The main intermediates during the decomposition of RDX contributed to the production of nitrogen gas. The concentration of nitrogen gas gradually increases, reaching 15 molecules at 14 ps after the initiation of RDX, then decreases 12 molecules during the whole simulation time. Among the five products, N2O is the most unstable one and its mean lifetime survived in the system is about 11.8 ps. N2O is induced by the hydrogen radical to form N2. The first CO2 was released at about 1.9 ps. The variation tendency for the concentration of CO2 is stable at the whole decomposition stage. CO2 is found to be in significantly smaller amounts than other species described above. Although many C—O bonds are formed, no more CO2 is generated in the subsequent simulations. At the beginning of the simulation, the O—H fragment is formed. Then the concentration of O—H increases to 17 molecules. O—H is induced by the hydrogen radical to form H2O. The concentration of H increases at the maximum of 4 molecules. H2O forms fast at the beginning and the number of H2O reaches the maximum of 10 molecules at 6 ps. Then the concentration H2O quickly decreases and reaches stable after 12 ps.
4 Conclusions(1) The decomposition of α-RDX at 3000 K coupled with 34.5 GPa is triggered by the homolysis of the C—H bond.
(2) The HONO elimination reaction during the decomposition is observed.
(3) The primary reactions of producing NO2, NO, N2O, N2, HONO, N2O4, H, O—H, H2O, and CO2 occur at very early stages
(4) After the initiation of RDX, there are three different decomposition pathways, their decomposition processes can be described by Fig. 4 shown in the text.
| [1] |
Isayev O, Gorb L, Qasim M, et al. Ab initio molecular dynamics study on the initial chemical events in nitramines: thermal decomposition of CL-20[J]. The Journal of Physical Chemistry B, 2008, 112(35): 11005-11013. DOI:10.1021/jp804765m |
| [2] |
Wu Q, Zhu W H, Xiao H M. An ab initio molecular dynamics study of thermal decomposition of 3, 6-di(azido)-1, 2, 4, 5-tetrazine[J]. Physical Chemistry Chemical Physics, 2014, 16(39): 21620-21628. DOI:10.1039/C4CP02579B |
| [3] |
Wu Q, Xiong G L, Zhu W H, et al. How does low temperature coupled with different pressures affect initiation mechanisms and subsequent decompositions in nitramine explosive HMX?[J]. Physical Chemistry Chemical Physics, 2015, 17(35): 22823-22831. DOI:10.1039/C5CP03257A |
| [4] |
Wu Q, Zhu W H, Xi ao, H M. Cooperative effects of different temperatures and pressures on the initial and subsequent decomposition reactions of the nitrogen-rich energetic crystal 3, 3'-dinitroamino-4, 4'-azoxyfurazan[J]. Physical Chemistry Chemical Physics, 2016, 18(10): 7093-7099. DOI:10.1039/C6CP00096G |
| [5] |
Oyumi Y, Brill T B. Thermal decomposition of energetic materials 3. A high-rate, in situ, FTIR study of the thermolysis of RDX and HMX with pressure and heating rate as variables[J]. Combustion Flame, 1985, 62(3): 213-224. DOI:10.1016/0010-2180(85)90147-6 |
| [6] |
Behrens R, Bulusu S. Thermal decomposition of energetic materials. Temporal behaviors of the rates of formation of the gaseous pyrolysis products from condensed-phase decomposition of 1, 3, 5-trinitrohexahydro-s-triazine[J]. The Journal of Physical Chemistry, 1992, 96(22): 8877-8891. DOI:10.1021/j100201a036 |
| [7] |
Behrens R, Bulusu S. Thermal decomposition of energetic materials. 4. Deuterium isotope effects and isotopic scrambling (HID, 13C/18O, l4N/l5N) in condensed-phase decomposition of 1, 3, 5-trkritrohexahydro-s-triazine[J]. The Journal of Physical Chemistry, 1992, 96(22): 8891-8897. DOI:10.1021/j100201a037 |
| [8] |
Hoffsommer J C, Glover D J. Thermal decomposition of 1, 3, 5-trinitro-1, 3, 5-triazacyclohexane (RDX): Kinetics of nitroso intermediates formation[J]. Combustion Flame, 1985, 59(3): 303-310. DOI:10.1016/0010-2180(85)90133-6 |
| [9] |
Bradley J N, Butler A K, Capey W D, et al. Mass spectrometric study of the thermal decomposition of 1, 3, 5-trinitrohexahydro-1, 3, 5-triazine (RDX)[J]. Journal of the Chemical Society, Faraday Transactions, 1977, 73(12): 1789-1795. |
| [10] |
Cosgrove J D, Owen A J. The thermal decomposition of 1, 3, 5 trinitro hexahydro-1, 3, 5 triazine (RDX)-part Ⅰ: The products and physical parameters[J]. Combustion Flame, 1974, 22(1): 13-18. DOI:10.1016/0010-2180(74)90004-2 |
| [11] |
Botcher T R, Wight C A. Explosive Thermal Decomposition Mechanism of RDX[J]. The Journal of Physical Chemistry, 1994, 98(21): 5441-5444. DOI:10.1021/j100072a009 |
| [12] |
Piermarini G J, Block S, Miller P J. Effects of pressure and temperature on the thermal decomposition rate and reaction mechanism of P-octa-hydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine[J]. The Journal of Physical Chemistry, 1987, 91(14): 3872-3878. DOI:10.1021/j100298a028 |
| [13] |
Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review A, 1965, 140(4): 1133-1138. |
| [14] |
Hohenberg P, Kohn W. Inhomogeneous electron gas[J]. Physical Review B, 1964, 136(3): 864-873. |
| [15] |
Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B: Condensed Matter, 1990, 41(11): 7892-7895. DOI:10.1103/PhysRevB.41.7892 |
| [16] |
Segall M D, Lindan P J D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. The Journal of Physical: Condensed Matter, 2002, 14(11): 2717-2744. DOI:10.1088/0953-8984/14/11/301 |
| [17] |
Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical Review Letter, 1996, 77(18): 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [18] |
Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J]. The Journal of Computational Chemistry, 2006, 27(15): 1787-1799. DOI:10.1002/(ISSN)1096-987X |
| [19] |
Tkatchenko A, Scheffler M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data[J]. Physical Review Letter, 2009, 102(7): 073005-073009. DOI:10.1103/PhysRevLett.102.073005 |
| [20] |
McCrone W C. RDX (Cyclotrimethylenetrinitramine)[J]. Analytical Chemistry, 1950, 22(7): 954-955. DOI:10.1021/ac60043a040 |
| [21] |
Car R, Parrinello M. Structural, dymanical, and electronic properties of amorphous silicon: An ab initio molecular-dynamics study[J]. Physical Review Letter, 1988, 60(3): 204-222. DOI:10.1103/PhysRevLett.60.204 |
| [22] |
Martyna G J, Tuckerman M E. A reciprocal space based method for treating long range interactions in ab initio and force-field-based calculations in clusters[J]. The Journal of Physical Chemistry, 1999, 110(6): 2810-2821. DOI:10.1063/1.477923 |
| [23] |
Zhao X, Hintsa E J, Lee Y T. Infrared multiphoton dissociation of RDX in a molecular beam[J]. The Journal of Physical Chemistry, 1988, 88(2): 801-810. DOI:10.1063/1.454158 |
| [24] |
Botcher T R, Wight C A. Explosive thermal decomposition mechanism of RDX[J]. The Journal of Physical Chemistry, 1994, 98(21): 5441-5444. DOI:10.1021/j100072a009 |
| [25] |
Capellos C, Papagiannakopoulos P, Liang Y L. The 248 nm photodecomposition of hexahydro-1, 3, 5-trinitro-1, 3, 5-triazine[J]. Chemistry Physical Letter, 1989, 164(5): 533-538. DOI:10.1016/0009-2614(89)85252-2 |
| [26] |
Weslowski S S, Fermann J T, Crawford T D, et al. The weakly bound dinitrogen tetroxide molecule: High level single reference wavefunctions are good enough[J]. The Journal of Chemistry Physical, 1997, 106(17): 7178-7184. DOI:10.1063/1.473679 |
| [27] |
Kovacs A, Borisenko K B, Pongor G. An application of the DFT-based scaled quantum mechanical force field method to a weakly bonded system: N2O4[J]. Chemistry Physical Letter, 1997, 280(5-6): 451-458. |
| [28] |
Jursic B S. A study of nitrogen oxides by using density functional theory and their comparison with ab initio and experimental data[J]. International Journal of Quantum Chemistry, 1996, 58(1): 41-46. DOI:10.1002/(ISSN)1097-461X |
| [29] |
Messerschmidt M, Wagner A, Wong M W, et al. Atomic properties of N2O4 based on its experimental charge density[J]. Journal of the American Chemical Society, 2002, 124(5): 732-733. DOI:10.1021/ja011802c |
The complete decomposition process of α-RDX crystal at 3000 K coupled with 34.5 GPa was studied via. ab initio MD simulation method.