



2. 氟氮化工资源高效开发与利用国家重点实验室, 陕西 西安 710065;
3. 陕西师范大学化学化工学院, 陕西 西安 710119
2. State Key Laboratory of Fluorine & Nitrogen Chemical, Xi′an 710065, China;
3. School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi′an 710119, China
1, 1-二氨基-2, 2-二硝基乙烯(FOX-7)是一种新型不敏感含能材料, 具有密度大、感度低、能量高等优点, 在不敏感武器装备中有广阔的应用前景, 受到各国研究人员的广泛关注[1-4]。对于含能材料而言, 其结晶形貌对安全性能和配方装填方式有着重要的影响[5]。因此, 深入研究FOX-7的结晶机制和形貌控制技术, 对于FOX-7的工程化制备有重要价值。
分子动力学方法可以模拟溶剂分子对特定晶面的吸附作用, 从分子层面对目标化合物的结晶过程和形貌实现高效、准确的分析和预测, 已广泛用于炸药分子在溶剂中结晶形貌的模拟, 如环四亚甲基四硝胺(HMX)、环三亚甲基三硝胺(RDX)、3, 4-二硝基呋咱基氧化呋咱(DNTF)及2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物(ANPyO)等[6-10]。任晓婷等[11]模拟了FOX-7在DMF和DMF/丙酮溶液中的晶体形貌, 结果显示DMF/丙酮溶液中FOX-7的生长晶形规则, 长径比小。赵强莉等[12]预测了FOX-7在多种溶剂中的晶体形貌, 分析了溶剂分子对FOX-7晶体生长的影响情况。本课题组采用修正的DREIDING力场对FOX-7的晶体形貌进行了模拟分析[13]。但上述工作仅研究了溶剂种类对FOX-7晶习的影响, 而忽略了结晶温度对其晶习的影响, 模拟准确性有待进一步提升。
本工作用分子动力学(MD)方法模拟了FOX-7的真空晶体形貌, 确定形态学重要生长面和附着能。进而通过构建溶剂-晶面模型, 使用修正附着能(MAE)模拟方法, 计算FOX-7主要晶面与溶剂分子的相互作用能。预测了FOX-7在不同温度下、不同比例的H2O/DMF溶剂中的结晶形貌, 并与文献实验结果比较验证了预测的准确性。预测结果可为FOX-7的制备和工艺放大提供参考。
2 计算理论及计算方法 2.1 计算理论附着能(AE)模型由Hartman和Bennema在基于周期性键链(PBC)理论[14-15]的基础上提出。根据该理论, 晶体表面成键所需时间与键合能成反比, 晶面生长速度与键合能成正比, 晶体生长最快的方向即为化学键最强的方向。AE模型在此基础上定义晶片能Eslice为生长出一层厚度为dhkl的晶片释放出的能量, 附着能Eatt为晶片附着在晶习表面(h k l)所释放出的能量, 两者之和即为晶体的晶格能Elatt[16]。
| $ {E_{{\rm{latt}}}} = {E_{{\rm{att}}}} + {E_{{\rm{slice}}}} $ | (1) |
通常情况下晶体由溶剂中生长得到, 如果晶面和溶剂间的吸附作用力较强, 溶剂分子在晶体界面会形成溶液表面层状结构。而晶面的生长需要吸附的溶剂分子离开晶面, 该去溶剂化过程需要耗费能量。不同晶面移除溶剂层所消耗能量不同, 其附着能变化也不同, 最终导致其在不同溶剂中的形貌发生变化。定义Eint为溶剂层与晶面间的相互作用能, Etot为溶剂层与晶面的总能量, Esurf和Esolv分别为晶面层和溶剂层的能量, 则:
| $ {E_{{\rm{int}}}} = {E_{{\rm{tot}}}} - {E_{{\rm{surf}}}} - {E_{{\rm{solv}}}} $ | (2) |
为了描述溶剂效应的影响, 对真空附着能Eatt引入修正项Es。Es代表溶剂与晶习表面(h k l)之间的结合能, 由式(3)计算得出, 其中Aacc为单晶胞晶面中的溶剂可接触面积, Abox为模型(h k l)面上的总晶面面积。最终经溶剂效应修正过的附着能(E′att)由式(4)计算得出。
| $ {E_{\rm{s}}} = {E_{{\rm{int}}}} \times \frac{{{A_{{\rm{acc}}}}}}{{{A_{{\rm{box}}}}}}{\rm{ }} $ | (3) |
| $ E{\prime _{{\rm{att}}}} = {E_{{\rm{att}}}} - {E_{\rm{s}}} $ | (4) |
使用Materials Studio 5.5软件[17], 根据FOX-7的单晶衍射数据[18]构建晶胞。FOX-7属于单斜晶系, P21/n空间群, α=γ=90.00°, β=90.61°, a=0.694 nm, b=0.664 nm, c=1.134 nm, 每个原胞中含有4个FOX-7分子。FOX-7的分子和晶胞结构如图 1所示。FOX-7晶胞经几何优化后, 在真空条件下使用AE模型模拟其晶体形貌, 获得形态学重要晶面(h k l)。为保持计算体系的一致性并提高运算速度, 进一步将原胞扩建为3×3×3的超晶胞并去掉周期性结构进行计算, 沿(0 1 1)、(1 0 -1)、(1 0 1)、(1 1 -1)、(0 0 2)和(1 1 0)六个主要晶面进行切割。选择三种不同比例的H2O/DMF混合溶液作为溶剂体系, H2O与DMF质量比分别为3:1, 1:1和1:3, 依次定义为溶剂Ⅰ、溶剂Ⅱ和溶剂Ⅲ。溶剂层由Amorphous Cell模块构建, 包含200个溶剂分子, 溶剂层盒子的长和宽与对应晶面的数值相符合。溶剂层经几何优化后, 在298 K下进行500 ps的NVT系综MD模拟使之达到平衡状态。将晶面层沿a/b/c方向固定作为双层结构模型的下层, 溶剂层作为双层结构模型的上层, 建模得到相应的动力学界面作用计算模型。为消除片层之间的相互作用, 在溶剂层上加一较厚的真空层(3 nm)。对该初始双层模型进行结构优化, 然后在NVT系综下(温度298 K, 控温Andersen thermostat)进行分子动力学模拟, 时间为1000 ps, 步长为1.0 fs, 以期达到平衡。待体系达到平衡之后, 继续进行动力学模拟过程, 时间设为500 ps, 步长为1.0 fs, 且每100个时间步长进行数据收集。对于势能的计算, 分别用Atom based和Ewald方法计算范德华和静电作用, 截断半径1.25 nm。最终取动力学轨迹中最后500帧计算体系的吸附能。晶面上溶剂的可及面积通过Connolly表面模型确定。

|
图 1 FOX-7的分子和晶胞结构图 Fig.1 The molecular and unit cell structure of FOX-7 |
COMPASS力场是一类基于从头计算(ab initio)的力场[19]。该力场不但能够模拟孤立分子的结构、振动频率、热力学性质等, 重要的是它能够模拟出更准确的凝聚态的结构与性质。首先通过结构优化及晶格能的计算验证该力场对FOX-7晶体的适用性。根据FOX-7晶体(α型)的单晶衍射实验数据建立3×3×3的超晶胞并对晶胞结构进行优化(COMPASS力场, Smart algorithm), 优化后的晶胞参数如表 1所示。可以看出, 优化所得晶胞参数与实验值基本吻合, 相对偏差除参数a以外不超过5%。此外, 在COMPASS力场下计算FOX-7的晶格能为-107.60 kJ·mol-1, 而实验测得FOX-7晶体升华热为108.78 kJ·mol-1, 所得晶格能为-113.74 kJ·mol-1[20], 与理论值十分接近。因此可认为COMPASS力场和3×3×3超晶胞模型具有较好的适用性。
| 表 1 FOX-7优化后的晶胞参数与实验值的比较 Tab.1 The comparison of the experimental and optimized lattice parameters of FOX-7 |
图 2所示为使用AE模型计算获得的FOX-7在真空条件下的晶体形貌, 其重要生长面及参数列于表 2。结果可知, FOX-7的真空晶习为纺锤形, 长径比为1.541, 其形态学重要生长面为(0 1 1), (1 0 -1), (1 0 1), (1 1 -1), (0 0 2)和(1 1 0)面。其中多重度为4的(0 1 1)面拥有最大的晶面面积(44.60%), 相应的附着能为-111.64 kJ·mol-1, 包括范德华力(-102.40 kJ·mol-1)和氢键作用(-9.24 kJ·mol-1)。因此(0 1 1)面为影响FOX-7晶体形态最重要的晶面。而(0 0 2)和(1 1 0)面占总面积比例分别仅为5.43%和3.45%, 趋于消失。

|
图 2 真空中FOX-7的晶习预测图 Fig.2 The predicted morphology of FOX-7 in vacuum |
| 表 2 真空中FOX-7重要晶面及参数 Tab.2 The crystal habit parameters of FOX-7 crystal in vacuum |
FOX-7的六个主要晶面与溶剂Ⅱ分子吸附界面模型达到热力学平衡后, 所得的平衡构型如图 3所示。可以看出, 溶剂分子已经紧密地附着在FOX-7晶面并形成了表面层状结构, 这说明FOX-7晶面和溶剂分子之间具有很强的吸附作用。混合溶剂中的DMF分子与晶面的作用力更强, 因此较H2O分子更贴近晶面。FOX-7各晶面均存在明显暴露的强极性基团(氨基或硝基基团), 故这些晶面皆为极性面, 而H2O和DMF分子均为极性分子, 因此有利于溶剂分子的吸附。FOX-7分子不是中心对称的生长单元, 其表面分子堆积取向呈现出多样性和复杂性的特点, 这就使得溶剂分子在各个晶面的吸附行为差异化。其中(1 0 -1)、(0 1 1)、(1 0 1)面上分子的排布相对比较平坦, 接触吸附相对不易; 而其他平面则凹凸不平, 表面有较多大空隙, 有利于溶剂分子的吸附。

|
图 3 FOX-7晶面-溶剂Ⅱ分子吸附模型的分子动力学平衡构型图 Fig.3 The configurations of FOX-7 surface-solvent Ⅱ interfaces from the MD equilibrium |
溶剂分子通过扩散作用吸附在FOX-7晶面上, 由于晶面生长时需消耗能量排除表面的溶剂分子, 使得附着能发生变化, 影响了晶体的最终形貌。表 3列出了溶剂Ⅱ-晶面双层结构模型的相互作用能、修正附着能及相对生长速率情况。相互作用能为负值表明两种界面具有相互吸引作用, 该作用越强则相互作用能的绝对值越大。计算可知在温度为298 K时, (0 1 1)面拥有最大的相互作用能(-1213.8 kJ·mol-1), 而(1 1 0)面的相互作用能(-435.2 kJ·mol-1)最小, 溶剂分子层对不同晶面吸附作用的大小依次为(0 1 1)>(1 1 -1)>(0 0 2)>(1 0 1)>(1 0 -1)>(1 1 0)。结果表明(0 1 1)和(1 1 -1)面对于溶剂分子的吸附能力较强, 而(1 0 -1)和(1 1 0)面由于相互作用能较小导致对溶剂分子的吸附能力弱。此外温度变化对相互作用能也有显著改变。当温度降至278 K时, (1 0 -1)面的相互作用能有了明显提高, (0 1 1)和(1 1 0)面的相互作用能则显著降低; 当温度升至318 K时, 除(1 0 1)和(1 1 0)面的相互作用能有明显变化以外, 其它晶面变化较小。
| 表 3 不同温度下溶剂Ⅱ和FOX-7主要晶面之间的相互作用能、修正附着能和相对生长速率 Tab.3 Calculated interaction energies, modified attachment energies and relative growth rates of FOX-7 crystal habit faces in solvent Ⅱ at different temperatures |
由表 3可知, 使用溶剂Ⅱ在温度为298 K时, (1 1 -1)面的修正附着能(10.84 kJ·mol-1)绝对值最小, 各晶面的修正附着能大小依次为(0 0 2)>(1 1 0)>(1 0 -1)>(0 1 1)>(1 0 1)>(1 1 -1)。因此(1 1 -1)面为形态学最重要晶面, 拥有最大的显露面积且相对生长速率最慢; 而(0 0 2)等四个面由于过快的生长速率导致消失。MAE模型预测的298 K下FOX-7在溶剂Ⅱ中的晶体形貌如图 4b所示, 为长方体状; 周群等[6]实验报道FOX-7在H2O/DMF溶剂中重结晶后的晶习如图 5所示[6], 对比可知理论预测与实验所得结果一致。其主要晶面为(1 1 -1)和(1 0 1)面, 其表面积分别占总面积的85.2%和14.8%。此外分析图 4可知, 在溶剂Ⅱ中当温度降低时, FOX-7晶形趋向于块状, 而温度升高时晶形趋向于片状。说明随温度条件变化FOX-7在溶液Ⅱ中的晶体形貌有明显差异。

|
图 4 FOX-7在溶剂Ⅱ中的预测晶体形貌 Fig.4 The predicted crystal morphologies of FOX-7 in solvent Ⅱ |

|
图 5 FOX-7在H2O/DMF溶剂中重结晶后的晶体形貌[6] Fig.5 The experimental crystal morphology of FOX-7 in H2O/DMF solvent |
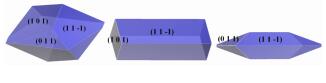
利用上述计算方法, 进一步研究了不同比例H2O/DMF溶剂对FOX-7晶体形貌的影响。利用MAE模型预测FOX-7在溶剂Ⅰ和溶剂Ⅲ中的晶体形貌如图 6和图 7所示。可以看出, 当温度为298 K时, FOX-7在溶剂Ⅰ和溶剂Ⅲ中的预测晶形均为梭形, 但主要生长面存在差异, 分别为(0 1 1)、(1 0 1)面和(1 1 -1)、(0 1 1)面。当温度降至278 K时, 溶剂Ⅰ中所得晶形为片状, 溶剂Ⅲ中所得晶形为长方体状; 当温度升至318 K时, 溶剂Ⅰ中所得晶形为椭球状, 溶剂Ⅲ中所得晶形与溶剂Ⅱ中相似, 为片状。在所有模拟条件下, FOX-7的(0 0 2)面均具有较大的修正附着能, 法向生长速度快, 故该面趋于消失。总体而言, FOX-7在298 K时不同比例H2O/DMF溶液中的晶体形貌多为规整的块状或梭形, 在受到机械刺激时不易破碎或折断, 显示出较低的感度, 故室温条件下制备的FOX-7晶体有更高的应用价值。

|
图 6 FOX-7在溶剂Ⅰ中的预测晶体形貌 Fig.6 The predicted crystal morphologies of FOX-7 in solvent Ⅰ |

|
图 7 FOX-7在溶剂Ⅲ中的预测晶体形貌 Fig.7 The predicted crystal morphologies of FOX-7 in solvent Ⅲ |
进一步选择溶剂Ⅱ体系, 在298 K时对动力学平衡后的轨迹进行径向分布函数(RDF)分析, 研究溶剂分子与FOX-7晶面的作用力方式和本质。图 8所示为FOX-7的(1 1 -1)晶面与DMF溶剂分子的径向分布函数计算结果。溶剂分子和晶面之间的分子间相互作用分为三种类型, 包括氢键(< 0.31 nm)、范德华力(0.31~0.50 nm)和静电力(>0.50 nm), 其中氢键和范德华力属于短程作用力, 静电力属于长程作用力。图 8a所示为DMF的氢原子和FOX-7的氧、氮原子之间的RDF分析结果。对于DMF的氢原子和FOX-7的氧原子之间的RDF分析显示, 在r=0.26 nm处的存在强尖峰, 可知二者之间存在很强的氢键; 而分析DMF的氢原子和FOX-7的氮原子之间的RDF, 在r<0.60 nm时无明显峰值, 在0.60~0.70 nm有强峰出现, 说明二者之间无氢键作用, 以静电作用力为主。图 8b所示为FOX-7的氢原子和DMF的氧、氮原子之间的RDF分析结果。对于FOX-7的氢原子和DMF的氧原子之间的RDF分析显示, 在r=0.28 nm和r=0.45 nm处分别有弱峰出现, 代表存在着较弱的氢键和范德华力, r=0.90 nm处的强峰说明静电作用力占主导地位; 而DMF的氢原子和FOX-7的氮原子之间仅在0.80~0.90 nm处出现强峰, 说明二者之间几乎无氢键和范德华力作用, 仅存在静电作用力。结合图 8a与图 8b分析可知, FOX-7的(1 1 -1)晶面与DMF分子间同时存在多种分子间作用力, 不同原子对之间的作用力存在较大差异。

|
图 8 DMF分子与FOX-7的(1 1 -1)面吸附模型的径向分布函数 Fig.8 RDFs between DMF and FOX-7 atoms for the (1 1 -1) face adsorption systems |
(1) FOX-7在真空条件下的晶习为纺锤形, 主要生长面为(0 1 1), (1 0 -1), (1 0 1), (1 1 -1), (0 0 2)和(1 1 0)面, 长径比为1.541。
(2) H2O/DMF溶剂分子对于FOX-7各晶面的吸附能力存在差异, 导致溶剂条件下FOX-7各晶面的附着能发生变化, 不同比例溶剂和温度条件下FOX-7的晶体形貌有明显差异, 其中在298 K时的晶体形貌为规整的块状或梭形, 与文献实验结果相吻合。
(3) 径向分布函数计算表明FOX-7的(1 1 -1)晶面与DMF分子间的作用力包括氢键、范德华力和静电力, 其中FOX-7的氧原子与DMF的氢原子之间的氢键作用力较强。
| [1] |
Latypov N V, Bergman J, Langlet A, et al. Synthesis and reactions of 1, 1-diamino-2, 2-dinitroethylene[J].
Tetrahedron, 1998, 54(38): 11525-11536. DOI:10.1016/S0040-4020(98)00673-5 |
| [2] |
Asta G, Lou M, Lulu H, et al. Proposed mechanism of 1, 1-diamino-2, 2-dinitroethylene decomposition: a density functional theory study[J].
J Phys Chem A, 1999, 103(50): 11045-11051. DOI:10.1021/jp991794a |
| [3] |
周诚, 黄新萍, 周彦水, 等. FOX-7的晶体结构和热分解特性[J].
火炸药学报, 2007, 30(1): 60-62. ZHOU Cheng, HUANG Xin-ping, ZHOU Yan-shui, et al. Crystal structure and thermal decomposition of FOX-7[J]. Chinese Journal of Explosives and Propellants, 2007, 30(1): 60-62. |
| [4] |
付秋菠, 舒远杰, 黄奕刚, 等. 1, 1-二氨基-2, 2-二硝基乙烯的合成与性能研究[J].
有机化学, 2006, 26(10): 1409-1413. FU Qiu-bo, SHU Yuan-jie, HUANG Yi-gang, et al. Synthesis and properties of 1, 1-diamino-2, 2-dinitroethylene[J]. Chinese Journal of Organic Chemistry, 2006, 26(10): 1409-1413. DOI:10.3321/j.issn:0253-2786.2006.10.012 |
| [5] |
Manner V W, Tappan B C, Scott B L, et al. Crystal structure, packing analysis, and structural-sensitivity correlations of erythritol tetranitrate[J].
Cryst Growth Des, 2014(14): 6154-6160. |
| [6] |
周群, 陈智群, 郑朝民, 等. FOX-7晶体形貌对感度的影响[J].
火炸药学报, 2014, 37(5): 67-69. ZHOU Qun, CHEN Zhi-qun, ZHENG Chao-min, et al. Effect of morphology of FOX-7 crystals on its sensitivity[J]. Chinese Journal of Explosives and Propellants, 2014, 37(5): 67-69. |
| [7] |
Chen G, Xia M Z, Wu L, et al. Prediction of crystal morphology of cyclotrimethylene trinitramine in the solvent medium by computer simulation: a case of cyclohexanone solvent[J].
J Phys Chem A, 2014(118): 11471-11478. |
| [8] |
Chen G, Chen C Y, Xia M Z, et al. A study of the solvent effect on the crystal morphology of hexogen by means of molecular dynamics simulations[J].
RSC Adv, 2015(5): 25581-25589. |
| [9] |
Liu N, Li Y N, Zeman S, et al. Crystal morphology of 3, 4-bis(3-nitrofurazan-4-yl)furoxan (DNTF) in a solvent system: molecular dynamics simulation and sensitivity study[J].
Crystengcomm, 2016(18): 2843-2851. |
| [10] |
石文艳, 王风云, 夏明珠, 等. 2, 6-二氨基-3, 5-二硝基吡啶-1-氧化物晶体形貌的MD模拟[J].
含能材料, 2016, 24(1): 19-26. SHI Wen-yan, WANG Feng-yun, XIA Ming-zhu, et al. Molecular dynamics simulation on the crystal morphology of 2, 6-diamino-3, 5-dinitropydine-1-oxide[J]. Chinese Journal of energetic materials(Hanneng Cailiao), 2016, 24(1): 19-26. DOI:10.11943/j.issn.1006-9941.2016.01.003 |
| [11] |
任晓婷, 叶丹阳, 丁宁, 等. 溶剂效应对FOX-7晶体形貌影响的分子动力学模拟研究[J].
兵工学报, 2015, 36(2): 272-278. REN Xiao-ting, YE Dan-yang, DING Ning, et al. A molecular dynamics simulation of solvent effects on the crystal morphology of FOX-7[J]. Acta Armamentarii, 2015, 36(2): 272-278. |
| [12] |
Zhao Q L, Liu N, Wang B Z, et al. A study of solvent selectivity on the crystal morphology of FOX-7 via a modified attachment energy model[J].
RSC Adv, 2016(6): 59784-59793. |
| [13] |
刘宁, 王伯周, 舒远杰, 等. FOX-7结晶形貌的分子动力学模拟[J].
火炸药学报, 2016, 39(2): 40-44. LIU Ning, WANG Bo-zhou, SHU Yuan-jie, et al. Molecular dynamics simulation on crystal morphology of FOX-7[J]. Chinese Journal of Explosives and Propellants, 2016, 39(2): 40-44. |
| [14] |
Hartman P, Bennema P. The attachment energy as a habit controlling factor: Ⅰ. Theoretical considerations[J].
J Cryst Growth, 1980(49): 145-156. |
| [15] |
Hartman P. The attachment energy as a habit controlling factor: Ⅲ. Application to corundum[J].
J Cryst Growth, 1980(49): 166-170. |
| [16] |
Berkovitch-Yellin Z. Toward an ab initio derivation of crystal morphology[J].
J Am Chem Soc, 1985(107): 8239-8253. |
| [17] |
Material Studio 5. 5, Acceryls Inc., San Diego, 2010.
|
| [18] |
Gilardi R. CCDC 127539: Experimental Crystal Structure Determination[DB]. Cambridge Crystallographic Data Centre, Cambridge, UK, 1999.
|
| [19] |
Sun H. Compass: an ab initio force field optimized for condensed phase application, overview with detail on alkane and benzene compounds[J].
J Phys Chem B, 1998, 102: 7338-7364. DOI:10.1021/jp980939v |
| [20] |
Sorescu D C, Boatz J A, Thompson D L. Classical and quantum-mechanical studies of crystalline FOX-7(1, 1-diamino-2, 2-dinitroethylene)[J].
J Phys Chem A, 2001, 105: 5010-5021. DOI:10.1021/jp010289m |
The crystal morphologies of FOX-7 in H2O/DMF solvents at different temperatures were predicted by modified attachment energy (MEA) model. The intermolecular interactions of crystal surface and solvent molecules were also studied.