







- Online First |
- Articles in press |
- Current Issue |
- Special Articles |
- Archive
-
LI Tai-chuan, MEI Yu-xin, LI Lan, ZHANG Chao-yang, HUANG Xin
Online:June 01, 2026 DOI: 10.11943/CJEM2026103
Abstract:To reveal the mechanism by which oriented alignment of thermally conductive fillers enhances the thermal conductivity of polymer matrix composites, a two-dimensional steady-state heat conduction numerical model was established for graphene/fluoropolymer composites. The effects of filler volume fraction, aspect ratio, and orientation angle on the effective thermal conductivity of the composites were systematically investigated. The results show that the effective thermal conductivity of the composite increases with the rise of graphene volume fraction and aspect ratio, but decreases with the increase of orientation angle. Graphene orientation exhibits a significant regulatory effect on directional thermal conductivity, and there is a cosine relationship between orientation angle and effective thermal conductivity, with the average coefficient of determination R2 of the fitting equation exceeding 0.99. At a volume fraction of 30% and an aspect ratio of 20∶1, as the orientation angle decreases from 90° to 10°, the effective thermal conductivity of the composite increases from 0.233 W·m-1·K-1 to 1.285 W·m-1·K-1, representing an increase of approximately 450%. This study demonstrates that oriented alignment of thermally conductive fillers can optimize the geometric matching between fillers and heat flow direction, improving the continuity and directionality of internal heat conduction pathways in composites, thereby significantly enhancing thermal transport capability along the target direction. The results can provide a theoretical basis for the structural design and performance regulation of thermally anisotropic high-thermal-conductivity composites.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
-
HE Yi-fei, ZHANG Hui, YIN Ping, PANG Si-ping
Online:May 29, 2026 DOI: 10.11943/CJEM2026025
Abstract:A novel dinitramine compound, N,N’-[5,5’-bis(trinitromethyl)-3,3’-bi-1,2,4-triazol]dinitramide (3), was synthesized via N-amination and subsequent nitration of the starting material 3,3’-bi(1H,1’H-1,2,4-triazole)-5,5’-bis(trinitromethyl) (1). The structure of compound 3 was confirmed by X-ray single-crystal diffraction (XRD) and further characterized by infrared spectroscopy (IR), elemental analysis (EA), and nuclear magnetic resonance (NMR) spectroscopy. The enthalpy of formation for compound 3 was calculated using the isodesmic reaction method, while its detonation performance was predicted using the EXPLO5 code. Results indicate that compound 3 crystallizes in the monoclinic space group P21/n. The crystal density of the dihydrate (3•2H2O) is 1.937 g•cm-3 at 100 K. It exhibits an initial decomposition temperature of 92.6 ℃ and a calculated enthalpy of formation of 680.5 kJ•mol-1. Furthermore, its predicted detonation velocity and pressure are 8926 m•s-1 and 34.1 GPa, respectively, with a specific impulse of 257.7 s. Its integrated performance suggests its potential for use as an energetic oxidizer.
- 1
- 2
- 3
- 4
- 5
-
ZHANG Jing, DOU Jin-kang, TANG Chang-wei, GAO Min, TAN Bo-jun, LIU Ning
Online:May 07, 2026 DOI: 10.11943/CJEM2026039
Abstract:To investigate the regulatory mechanism of amino and nitro substituents on the thermal stability of TYX series heat-resistant explosives, two novel heat-resistant explosives based on the bis(triazolo)tetrazine backbone-fully amino-substituted 2,7-diaminobis([1,2,4]triazolo)[1,5-b:1',5'-e][1,2,4,5]tetrazine-5,10-diium-3,8-diide (TYX-1) and mono-nitro mono-amino substituted 2-amino-7-nitrobis([1,2,4]triazolo)[1,5-b:1',5'-e][1,2,4,5]tetrazine-5,10-diium-3,8-diide (TYX-3)-were selected in this study. Their thermal decomposition behaviors were systematically compared using differential scanning calorimetry (DSC) and thermal decomposition kinetic methods, while the decomposition processes were comprehensively analyzed by thermogravimetry-infrared-mass spectrometry (TG-FTIR-MS). The results show that the difference in substituents exerts a decisive influence on their thermal stability and decomposition pathways. TYX-1 exhibits a single high-temperature decomposition process with a peak temperature of 477.56 ℃ (at a heating rate of 20 ℃·min-1), and its decomposition mechanism conforms to the random two-dimensional nucleation growth model (A2), consistent with the layered stacking structure promoted by amino groups and the resulting controlled two-dimensional energy release pathway. In contrast, TYX-3 shows a significantly lower decomposition temperature and multi-step decomposition characteristics: the initial stage follows a two-dimensional diffusion model (D2), followed by a multi-reaction competitive stage, and finally transitions to a skeletal fracture process dominated by the one-dimensional chemical reaction model (F1). Gas product analysis shows that the main decomposition products of TYX-1 are N2, CO2, N2O, and HCN, while additional products including NO, HCNO, NH2, and H2O are detected for TYX-3, confirming that the nitro group, as a strong oxidizing moiety, induces an unconventional decomposition pathway and promotes the oxidative cleavage of the parent ring skeleton.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
-
LIU Song-yuan, XIA Yu, CHENG Zhi-peng, LV Jia-lu, LEI Kang, LIANG Jian-hao, WU Xing-liang, XU Sen
Online:April 29, 2026 DOI: 10.11943/CJEM2026060
Abstract:To investigate the evolution of the mechanical sensitivity of the energetic oxidizer ammonium dinitramide (ADN) under varying moisture and temperature conditions, standard tests specified by the Federal Institute for Materials Research and Testing (BAM) were carried out in combination with the Langlie–D optimization method. The impact and friction sensitivities of ADN samples with moisture contents of 0, 5%, 10%, and 15% were quantitatively evaluated at 25, 50, and 75 ℃. The results show that both impact and friction sensitivities decrease significantly with increasing moisture content, although the variation is not simply linear and instead exhibits a distinct stagewise pattern. At 25 ℃, the limiting impact energy increased from 4 J for anhydrous ADN to 30 J at 5% moisture content and exceeded 50 J when the moisture content reached 10% or higher. Meanwhile, the minimum friction load increased progressively from 56 N and surpassed the upper measurement limit of the apparatus at 15% moisture content. Further analysis indicates that increasing temperature gradually weakens the desensitizing effect of moisture. At a given moisture content, both the limiting impact energy and the minimum friction load at 50 and 75 ℃ are generally lower than those at 25 ℃, while the corresponding impact and friction ignition-probability curves shift continuously toward lower stimulus levels. Among them, friction sensitivity shows a stronger dependence on temperature. These results suggest that the coupled effects of moisture content and temperature are reflected not only in single threshold values, but also in systematic shifts of the entire ignition-probability curve and the corresponding low-probability risk boundary.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
-
TANG Chang-wei, HE Dan, GE Zhong-xue, HE Jin-feng, ZHANG Ru-xin, WANG Xiao-ying
Online:April 20, 2026 DOI: 10.11943/CJEM2026029
Abstract:The inherent conflict between high energy density and low mechanical sensitivity represents a central challenge in the field of energetic materials. Although traditional nitramine compounds such as RDX, HMX, and CL-20 have significantly enhanced energy levels, they remain constrained by this trade-off. To explore new pathways for overcoming conventional performance limitations, researchers have proposed a strategy centered on rigid, planar fused-ring frameworks, leading to the development of nitrogen-rich fused-ring compounds. This review systematically outlines the evolution of this field, from the design of monocyclic systems (e.g., azoles, azines, and 1,2,5-oxadiazoles) to the integrated design of binary fused-ring systems. It highlights the conceptual design of representative molecules, key advances in synthetic methodologies—ranging from oxidative nitration to controlled rearrangement reactions—and the regulatory mechanisms of intermolecular interactions such as hydrogen-bonding networks and π-π stacking on material performance. This progression illustrates a paradigm shift from empirical trial-and-error to rational design and function-oriented customization. Finally, addressing the synthetic bottlenecks that constrain practical application, this review proposes that future breakthroughs require synergistic efforts across three dimensions: design, preparation, and application. This includes developing design methods that balance performance with synthetic feasibility, promoting synthetic technologies with improved safety profiles, and expanding the application scope of fused-ring energetic materials, thereby facilitating the transition from molecular design to practical implementation and providing a foundation for next-generation high-performance energetic materials.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
- 105
- 106
- 107
- 108
- 109
- 110
- 111
- 112
- 113
- 114
- 115
- 116
- 117
- 118
- 119
- 120
- 121
- 122
- 123
-
GUO Hao-Yu, YU Yong-Gang, HU Yu-Bo, ZHANG Xin-Wei
Online:April 26, 2026 DOI: 10.11943/CJEM2026016
Abstract:To clarify the evolution mechanism of the gas curtain flow field during underwater launch and optimize the in-tube drainage efficiency, this study investigates the effects of different injection pressure conditions on the evolution characteristics of the gas curtain inside a spiral-grooved underwater gun tube. A transient three-dimensional two-phase flow model was established for the drainage process during underwater gas-curtain launch. Based on a 40 mm supercavitating projectile and a spiral-grooved gas-curtain launch tube, numerical simulations of gas curtain evolution were performed under four different injection pressure conditions, and the effect of the pressurization rate on key evolution characteristics was analyzed. The results show that a higher pressurization rate results in better overall drainage performance, but it also induces higher pressure ahead of the projectile. Specifically, as the pressurization rate increases from 1 MPa·ms-1 to 4 MPa·ms-1, the drainage completion time (i.e., the time required for the gas curtain front to reach the muzzle) decreases from 14.4 ms to 11.8 ms, achieving an 18.1% improvement in time efficiency and a 25.7% increase in drainage capacity. However, upon completion of drainage, the pressure on the projectile surface increases from 3.7 MPa to 4.96 MPa, and the average pressure inside the tube rises from 4.14 MPa to 5.47 MPa. In conclusion, although a high pressurization rate can significantly accelerate drainage, it substantially increases the subsequent in-tube motion resistance of the projectile. Therefore, in the practical matching design of propellant charge and projectile, it is necessary to comprehensively balance drainage efficiency and initial motion resistance, and reasonably control the injection pressure gradient.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
-
SHI Jun-hao, JIANG Tian-yu, ZHANG Wen-quan
Online:April 15, 2026 DOI: 10.11943/CJEM2026057
Abstract:Stereoisomerism plays a unique role in tuning the structures and performances of energetic molecules. Photochemical reactions feature mild conditions and precise configurational regulation thus bear important theoretical and practical significance for realizing stereoisomeric transformation of energetic molecules. (E)-Potassium 5,5'-azotetrazolate (E-PZT) was employed as the substrate in this work. Systematic condition screening was conducted to determine the optimal parameters for the photochemical synthesis of (Z)-potassium 5,5'-azotetrazolate (Z-PZT). The molecular structure of Z-PZT was fully characterized. The half-life of Z-PZT is measured to be 49 min at room-temperature. Theoretical calculations highly consistent with the experimental phenomena and results of the photoinduced isomerization reaction. The predicted excitation wavelengths and corresponding spectra obtained from theoretical calculations are highly consistent with the experimental phenomena and results of the photoinduced isomerization reaction. The successful preparation of the target product Z-PZT is further verified by these theoretical results. The isomerization energy barrier was calculated, and a rational photochemical reaction mechanism was accordingly proposed to elucidate the metastable characteristic of Z-PZT and the intrinsic origin of its relatively short half-life. Reliable experimental and theoretical support is provided by this work for the investigation of photoinduced isomerization regulation in energetic molecules.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
-
Online:April 08, 2026 DOI: 10.11943/CJEM2025257
Abstract:To address the issue of significant attenuation and strong scattering of ultrasonic waves in solid propellants, which prevents existing ultrasonic transducers from detecting all internal defects, a nondestructive testing research method using ultrasonic laminated transducers is proposed. Based on the research of domestic and international scholars, this study first proposes leveraging the high transmission energy characteristic of laminated transducers to mitigate attenuation and scattering problems during ultrasonic propagation in solid rocket propellants. A 1 MHz four-layer ultrasonic transducer is designed. This transducer converts a planar acoustic field into a cylindrical acoustic field through crystal stacking, thereby enriching echo information and improving defect resolution. Based on acoustic field simulations of the ultrasonic laminated transducer, a 1 MHz four-layer ultrasonic transducer is developed. Experimental comparisons with domestic and international transducers of similar specifications show a 20 dB improvement in gain. Finally, an ultrasonic automated inspection system for solid rocket propellants is established, enabling online inspection of solid rocket propellants. The results indicate that the developed inspection equipment can accurately detect the smallest artificial blind holes measuring Φ1.2 mm × 5 mm (depth) as well as natural inclusion defects, achieving qualitative and quantitative nondestructive testing of all internal defects in solid rocket propellants.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
-
GONG Ru-nan, CHENG Guo-rong, ZHAO Zhuo-an, CHEN Song, LIU Suo-en, LIAN Jian-biao, ZHANG Bing, CHEN Jin-fang, LIU Xiao-lu
Online:April 07, 2026 DOI: 10.11943/CJEM2026005
Abstract:In order to explore the effect of aromatic compounds on the combustion performance of CMDB propellant, 2,2"-(propane-1,1-diyl)bis(4-(tert-butyl)phenol) (PDBP) was used to prepare HMX-CMDB propellant, and the combustion performance of HMX-CMDB propellants with different PDBP contents was studied using the target line method. The results showed that with the increase of PDBP content, the burning rate and pressure index of HMX-CMDB propellant significantly decreased. For HMX-CMDB propellant containing 7% PDBP, the burning rate at 16 MPa decreased to 8.55 mm·s-1, and the pressure index decreased to 0.217. Compared with the sucrose octaacetate (SOA), HMX-CMDB propellant containing the same mass of PDBP exhibited almost unchanged burning rate in the low-pressure region (8-10 MPa), a further reduced burning rate in the high-pressure region (12-16 MPa), resulting in a lower pressure index. In addition, after replacing SOA with 1% PDBP, the detonation heat of HMX-CMDB propellant was reduced by only 20 kJ/kg. Based on chemical structure analysis, the mechanism of PDBP"s burning rate inhibition effect is as follows: the aromatic molecule decompose to form protons and structurally stable radical molecules with conjugated π bonds, and the active protons can react with the active radicals released from the decomposition of energetic molecules in the HMX-CMDB propellant, forming stable structures that mitigate the autocatalytic effect of free radicals, thereby reducing the burning rate.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
-
LI Xu-han, XU Cong, LIU Ji-hong, ZHANG Yan
Online:April 13, 2026 DOI: 10.11943/CJEM2026010
Abstract:In order to meet the multiple requirements of high safety, low cost, high temperature resistance, and high pressure resistance on the seismic source for deep oil exploration, a kind of high-voltage switch consisting of two gas discharge tubes (GDTs) was used to control the discharge of thin film capacitor, and S-type bridge foil was exploded into high-temperature and high-pressure gas/plasma to ignite the boron potassium nitrate (BPN) pellet and generate underwater shock waves. Then, the ignition threshold of the seismic source was determined by the up-down method, and the acoustic characteristics were also studied using pressure probe. The results show that the firing unit composed of a thin film capacitor (2 μF) and the high-voltage switch based on GDTs can reliably achieve pulse discharge, and work at the extreme underground environments such as high temperature and high pressure, and the cost is less than ¥10. Using the firing unit to stimulate the S-type Cu bridge foil, a firing voltage threshold of 1100 V and a critical peak current of 1847 A were determined. The measurements of underwater shock waves and acoustic source level (ASL) analysis show that the ASL excited by the seismic source is higher than 150 dB in the 50-800 Hz frequency band, meeting the requirements of underground seismic sources.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
-
FANG-Song-hang, LUO Gui-ying, YOU Ting, DAI Jiu-shaung, ZHOU Jie
Online:March 31, 2026 DOI: 10.11943/CJEM2025248
Abstract:To improve the plasticizing effect of high-nitrogen single-base gun propellant and determine appropriate process parameters for high-nitrogen nitrocellulose (NC) during continuous twin-screw plastication, Molecular dynamics simulation was employed to analyze the effects of ethanol-ether mass ratio and solvent-NC mass ratio on NC plasticization. Ethanol-ether solubility experiments and rheological tests of the plasticized material were conducted to verify the simulation results. Results show that the solubility parameter of the ethanol-ether mixed solvent closely matches that of high-nitrogen NC. Strong hydrogen-bonding and electrostatic interactions exist between high-nitrogen NC and ethanol, while van der Waals forces dominate between NC and ether. At an ethanol-ether mass ratio of 1∶1.4, ethanol forms strong hydrogen bonds with NC, resulting in higher solubility, which is in good consistency with the experimental results that NC exhibits maximum solubility at an ethanol-ether mass ratio of 1∶1.36. Increasing the solvent-NC mass ratio within a certain range weakens the intramolecular hydrogen-bond interaction of NC and increases the radius of gyration of the molecular chains. These changes are correlated with the macroscopic phenomena of reduced shear viscosity of the material and a more compact and uniform extruded strands surface. At a solvent-to-NC mass ratio of 0.85, NC exhibits the largest radius of gyration for NC (2.24 nm) and the fewest intramolecular hydrogen bonds, which aligns with the experimental result that the apparent shear viscosity of the material is minimized at a solvent-NC mass ratio of 0.825. Due to the combined effects of strong screw shear and solvent volatilization, it is recommended to use a lower screw speed when plasticizing high-nitrogen NC with the simulated solvent-NC mass ratio. Additionally, appropriately increasing the screw speed under a low solvent-NC mass ratio can also effectively reduce material viscosity, attention must be given to the potential adverse effects caused by shear-induced heating.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
-
WANG Cheng, WEI Ding, LI Xing-han, WANG Hang-yu, LIN Jia-rui, CHEN Hao-dong, GAN Yun-dan
Online:April 03, 2026 DOI: 10.11943/CJEM2025269
Abstract:To elucidate the energy deposition mechanism of electro-chemical coupled explosion and provide a scientific basis for parameter optimization and precise control of related devices, an experimental platform was established to systematically investigate the effects of aluminum wire diameter (0.1-0.4 mm) and initial charging voltage (25-40 kV) on the detonation of HMX driven by electrical wire explosion. The results reveal that the electro-chemical coupled explosion comprises four characteristic stages: wire vaporization and plasma expansion, HMX ignition, HMX detonation, and disintegration of the detonation-product conductive channel. A quantitative criterion system for identifying mechanism transitions was established by defining the energy fraction in the HMX detonation stage (ηⅢ=EⅢ/Etotal) and the power peak ratio (γ=Pp2/Pp1). When ηⅢ > 90% and γ > 0.5, the system operates in the HMX-dominated “electro-chemical coupled explosion” mode; when ηⅢ decreases to 70%-80% and γ <0.2, it transitions to the Al-dominated “electrical explosion” mode; when ηⅢ≈0, γ≈0, and current oscillations disappear, it enters the resistance-dominated “capacitive discharge” mode. The wire diameter governs the fundamental transition of energy deposition mechanisms by controlling the effective vaporized and ionized mass fraction of aluminum. As the diameter increases from 0.1 mm to 0.4 mm, the energy release mechanism sequentially undergoes the three modes described above. The initial charging voltage regulates the intensity and efficiency of the coupled explosion through a power density enhancement mechanism. Increasing the voltage from 25 kV to 40 kV boosts the first power peak by 3.2 times, shortens the ignition delay by 62%, increases the energy deposited in the HMX detonation stage by 3.0 times, and raises the total deposited energy by 3.3 times. This study demonstrates that enhancing the efficiency of electro-chemical coupled explosion requires a combined strategy of reducing the wire diameter and increasing the initial charging voltage. The established quantitative criterion system and synergistic regulation laws provide a critical scientific basis for parameter optimization and precise control of electro-chemical coupled explosion technology.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
-
LIN Gao-ming, WANG Su-wei, LIU Xiao-lu, ZHU Lin-yi, LIU Yao, WANG Kang
Online:March 18, 2026 DOI: 10.11943/CJEM2025242
Abstract:To address the challenges of low automation, high danger, and poor uniformity in conventional charging processes of thermoplastic energetic materials, this study introduced a vertical screw charging technology and established a quantitative comprehensive performance evaluation method to guide process optimization, aiming to systematically enhance process efficiency, charging quality, and operational safety. Based on an analysis of the viscoelastic properties of the slurry, the rheological behavior of the slurry during extrusion under different process conditions and formulation components was simulated, and the formation mechanisms of high-temperature and high-pressure hot spots were investigated. The results show that increasing the solid content mass fraction from 75% to 85% significantly reduces the slurry flowability, with increases in flow field pressure and shear stress by 827% and 600%, respectively, and an increase in viscous heating by 384 kW·m-3. These changes intensify the thermo-mechanical coupling behavior during screw extrusion, reduce process safety, and raise the process risk coefficient from 0.99 to 3.36. However, by adjusting the screw speed (within the range of 10 r·min-1 to 30 r·min-1) and incorporating metal particles to enhance the thermal conduction network among the barrel, slurry, and screw, the temperature fluctuation range can be reduced by 0.8 ℃ to 1.9 ℃, effectively suppressing the formation of local hot spots.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
-
LIU Jin-ming, ZHAO Xin, LI Wei, E Xiu-tian-feng
Online:March 17, 2026 DOI: 10.11943/CJEM2026002
Abstract:To investigate the effect of low-molecule-weight gelators on the overall performance of gel fuels, five acylhydrazone-based low-molecular-weight gelators were designed and synthesized. Kerosene gel fuels were prepared using the heating-cooling method, and the gelation mechanism was examined. A comprehensive evaluation system for gelator performance was established based on six parameters: the minimum addition amount of gelators (A), the phase transition temperature (Tg), the physicochemical stability (Spc), the loss rate of energy density (Eloss), the shear thinning capacity (Sthin) and the resetting property (R). The results showed that all gelators could form three-dimensional network structures through non-covalent interactions such as hydrogen bonds and π-π conjugation, effectively confining kerosene molecules. Their gel fuels exhibited thermal reversibility (Tg = 50-80 ℃) and had good physical and chemical stability and shear thinning behavior. Among them, L18 gelator had the lowest minimum addition amount (3.1%) and the fastest gelation speed (15 s). L5 gelator had the best physical and chemical stability and the mass retention rate was 97.5% at a high centrifugal speed of 10000 r·min-1. L16 gelator had the strongest shear thinning ability, with a viscosity of only 34.72 mPa·s after shearing. The conclusion indicated that based on the multi-dimensional performance evaluation system established by the institute, L16 and L18 gelators demonstrated significant comprehensive advantages.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
-
YANG Li-jie, ZHAI Jin-xian, XING Zi-han, TONG Tian-lin
Online:March 12, 2026 DOI: 10.11943/CJEM2025270
Abstract:To reveal the curing reaction characteristics of polyurethane-crosslinked energetic composite slurry, this study derived the curing kinetic equations for the HTPB/IPDI system based on the evolution of characteristic functional groups monitored by in-situ ATR-FTIR spectroscopy during the curing process. The evolution of characteristic groups was monitored at 45 ℃, 50 ℃, 55 ℃, 60 ℃ and 65 ℃. Using the derived equations, kinetic curves for the slurry curing reaction were constructed, and the apparent activation energy was determined. The results indicate a two-stage curing process. The first stage corresponds to the pre-gelation period, with an apparent activation energy (Ea1) of 69.83±5.54 kJ·mol-1. The second stage corresponds to the post-gelation curing period, with an apparent activation energy (Ea2) of 71.31±4.45 kJ·mol-1. The apparent activation energy for the HTPB/IPDI energetic composite slurry is significantly higher than that of a homogeneous HTPB/IPDI mixture.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
-
MENG Xing, ZHEN Jiang-tao, XU Lin-nan, LI Jun
Online:March 09, 2026 DOI: 10.11943/CJEM2025255
Abstract:To achieve one-step determination of component contents in hydroxylammonium nitrate-based propellants (HAN-based propellant) and address issues like complex procedures and long analysis cycles of existing methods, molecular spectroscopy multivariate calibration technology was employed to develop the analytical method for such propellants. Near-infrared (NIR) spectra of prepared HAN-based propellant samples were acquired. Using partial least squares (PLS), the optimal spectral pretreatment methods, spectral regions, and number of principal components (NPC) were selected, and outliers were eliminated. Separate NIR quantitative analysis models were established for the four main components in the propellant: hydroxylammonium nitrate, nitrate A, additive B and additive C. All established models exhibit excellent performance. The correlation coefficients of the calibration set (RC) are all above 0.997, and those of the validation set (RP) are all above 0.990. The standard error of calibration (SEC) is below 0.06 for all models, and the standard error of prediction (SEP) is below 0.09. Additionally, the ratio of SEP to SEC is less than 2 for each model. The results show that the NIR method has good consistency with manual titration and gas chromatography (GC). The test deviations of the four components are all less than 0.10%. The NIR method also demonstrates high precision. The standard deviations (SD) of repeated tests for hydroxylammonium nitrate and nitrate A are less than 0.10%. For additive B and additive C, the SD of repeated tests are less than 0.03%. The established quantitative analysis models and method enable simple, rapid and one-step determination of component contents in HAN-based propellants.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
-
DOU Kai-le, ZHAO Wei-bo, HE Chun-lin, ZHANG Lei, PANG Si-ping
Online:February 03, 2026 DOI: 10.11943/CJEM2025237
Abstract:Energetic materials have attracted significant attention due to their critical roles in national defense, aerospace, and specialized engineering applications. However, their research and development are hindered by high experimental costs, safety risks, and lengthy synthesis cycles, which greatly limit the rapid iteration and practical deployment of novel energetic compounds. In recent years, machine learning (ML) has emerged as a powerful tool in chemistry and materials science owing to its strong capabilities in data modeling and prediction. This review summarizes the latest advances in machine learning–assisted chemical synthesis, focusing on three major aspects: reaction prediction, synthesis route planning, and automated synthesis. Particular emphasis is placed on the potential value and limitations of applying ML techniques to energetic material synthesis. The key challenges—such as data scarcity and inconsistency, lack of safety evaluation frameworks, and limited experimental validation and model retraining—are also discussed. Finally, the review outlines future perspectives, including the establishment of standardized and shareable databases, and the development of high-throughput and automated experimental platforms tailored for energetic systems. This work aims to provide theoretical insights and methodological support for achieving efficient and intelligent synthesis of energetic materials.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16

















































































































































































































































































































































































































































































































































































































































































































































-
LIU Jin-ming, ZHAO Xin, LI Wei, E Xiu-tian-feng
Online:March 17, 2026 DOI: 10.11943/CJEM2026002
Abstract:To investigate the effect of low-molecule-weight gelators on the overall performance of gel fuels, five acylhydrazone-based low-molecular-weight gelators were designed and synthesized. Kerosene gel fuels were prepared using the heating-cooling method, and the gelation mechanism was examined. A comprehensive evaluation system for gelator performance was established based on six parameters: the minimum addition amount of gelators (A), the phase transition temperature (Tg), the physicochemical stability (Spc), the loss rate of energy density (Eloss), the shear thinning capacity (Sthin) and the resetting property (R). The results showed that all gelators could form three-dimensional network structures through non-covalent interactions such as hydrogen bonds and π-π conjugation, effectively confining kerosene molecules. Their gel fuels exhibited thermal reversibility (Tg = 50-80 ℃) and had good physical and chemical stability and shear thinning behavior. Among them, L18 gelator had the lowest minimum addition amount (3.1%) and the fastest gelation speed (15 s). L5 gelator had the best physical and chemical stability and the mass retention rate was 97.5% at a high centrifugal speed of 10000 r·min-1. L16 gelator had the strongest shear thinning ability, with a viscosity of only 34.72 mPa·s after shearing. The conclusion indicated that based on the multi-dimensional performance evaluation system established by the institute, L16 and L18 gelators demonstrated significant comprehensive advantages.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
-
MENG Xing, ZHEN Jiang-tao, XU Lin-nan, LI Jun
Online:March 09, 2026 DOI: 10.11943/CJEM2025255
Abstract:To achieve one-step determination of component contents in hydroxylammonium nitrate-based propellants (HAN-based propellant) and address issues like complex procedures and long analysis cycles of existing methods, molecular spectroscopy multivariate calibration technology was employed to develop the analytical method for such propellants. Near-infrared (NIR) spectra of prepared HAN-based propellant samples were acquired. Using partial least squares (PLS), the optimal spectral pretreatment methods, spectral regions, and number of principal components (NPC) were selected, and outliers were eliminated. Separate NIR quantitative analysis models were established for the four main components in the propellant: hydroxylammonium nitrate, nitrate A, additive B and additive C. All established models exhibit excellent performance. The correlation coefficients of the calibration set (RC) are all above 0.997, and those of the validation set (RP) are all above 0.990. The standard error of calibration (SEC) is below 0.06 for all models, and the standard error of prediction (SEP) is below 0.09. Additionally, the ratio of SEP to SEC is less than 2 for each model. The results show that the NIR method has good consistency with manual titration and gas chromatography (GC). The test deviations of the four components are all less than 0.10%. The NIR method also demonstrates high precision. The standard deviations (SD) of repeated tests for hydroxylammonium nitrate and nitrate A are less than 0.10%. For additive B and additive C, the SD of repeated tests are less than 0.03%. The established quantitative analysis models and method enable simple, rapid and one-step determination of component contents in HAN-based propellants.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
-
YANG Li-jie, ZHAI Jin-xian, XING Zi-han, TONG Tian-lin
Online:March 12, 2026 DOI: 10.11943/CJEM2025270
Abstract:To reveal the curing reaction characteristics of polyurethane-crosslinked energetic composite slurry, this study derived the curing kinetic equations for the HTPB/IPDI system based on the evolution of characteristic functional groups monitored by in-situ ATR-FTIR spectroscopy during the curing process. The evolution of characteristic groups was monitored at 45 ℃, 50 ℃, 55 ℃, 60 ℃ and 65 ℃. Using the derived equations, kinetic curves for the slurry curing reaction were constructed, and the apparent activation energy was determined. The results indicate a two-stage curing process. The first stage corresponds to the pre-gelation period, with an apparent activation energy (Ea1) of 69.83±5.54 kJ·mol-1. The second stage corresponds to the post-gelation curing period, with an apparent activation energy (Ea2) of 71.31±4.45 kJ·mol-1. The apparent activation energy for the HTPB/IPDI energetic composite slurry is significantly higher than that of a homogeneous HTPB/IPDI mixture.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
-
WANG Cheng, WEI Ding, LI Xing-han, WANG Hang-yu, LIN Jia-rui, CHEN Hao-dong, GAN Yun-dan
Online:April 03, 2026 DOI: 10.11943/CJEM2025269
Abstract:To elucidate the energy deposition mechanism of electro-chemical coupled explosion and provide a scientific basis for parameter optimization and precise control of related devices, an experimental platform was established to systematically investigate the effects of aluminum wire diameter (0.1-0.4 mm) and initial charging voltage (25-40 kV) on the detonation of HMX driven by electrical wire explosion. The results reveal that the electro-chemical coupled explosion comprises four characteristic stages: wire vaporization and plasma expansion, HMX ignition, HMX detonation, and disintegration of the detonation-product conductive channel. A quantitative criterion system for identifying mechanism transitions was established by defining the energy fraction in the HMX detonation stage (ηⅢ=EⅢ/Etotal) and the power peak ratio (γ=Pp2/Pp1). When ηⅢ > 90% and γ > 0.5, the system operates in the HMX-dominated “electro-chemical coupled explosion” mode; when ηⅢ decreases to 70%-80% and γ <0.2, it transitions to the Al-dominated “electrical explosion” mode; when ηⅢ≈0, γ≈0, and current oscillations disappear, it enters the resistance-dominated “capacitive discharge” mode. The wire diameter governs the fundamental transition of energy deposition mechanisms by controlling the effective vaporized and ionized mass fraction of aluminum. As the diameter increases from 0.1 mm to 0.4 mm, the energy release mechanism sequentially undergoes the three modes described above. The initial charging voltage regulates the intensity and efficiency of the coupled explosion through a power density enhancement mechanism. Increasing the voltage from 25 kV to 40 kV boosts the first power peak by 3.2 times, shortens the ignition delay by 62%, increases the energy deposited in the HMX detonation stage by 3.0 times, and raises the total deposited energy by 3.3 times. This study demonstrates that enhancing the efficiency of electro-chemical coupled explosion requires a combined strategy of reducing the wire diameter and increasing the initial charging voltage. The established quantitative criterion system and synergistic regulation laws provide a critical scientific basis for parameter optimization and precise control of electro-chemical coupled explosion technology.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
-
GONG Ru-nan, CHENG Guo-rong, ZHAO Zhuo-an, CHEN Song, LIU Suo-en, LIAN Jian-biao, ZHANG Bing, CHEN Jin-fang, LIU Xiao-lu
Online:April 07, 2026 DOI: 10.11943/CJEM2026005
Abstract:In order to explore the effect of aromatic compounds on the combustion performance of CMDB propellant, 2,2"-(propane-1,1-diyl)bis(4-(tert-butyl)phenol) (PDBP) was used to prepare HMX-CMDB propellant, and the combustion performance of HMX-CMDB propellants with different PDBP contents was studied using the target line method. The results showed that with the increase of PDBP content, the burning rate and pressure index of HMX-CMDB propellant significantly decreased. For HMX-CMDB propellant containing 7% PDBP, the burning rate at 16 MPa decreased to 8.55 mm·s-1, and the pressure index decreased to 0.217. Compared with the sucrose octaacetate (SOA), HMX-CMDB propellant containing the same mass of PDBP exhibited almost unchanged burning rate in the low-pressure region (8-10 MPa), a further reduced burning rate in the high-pressure region (12-16 MPa), resulting in a lower pressure index. In addition, after replacing SOA with 1% PDBP, the detonation heat of HMX-CMDB propellant was reduced by only 20 kJ/kg. Based on chemical structure analysis, the mechanism of PDBP"s burning rate inhibition effect is as follows: the aromatic molecule decompose to form protons and structurally stable radical molecules with conjugated π bonds, and the active protons can react with the active radicals released from the decomposition of energetic molecules in the HMX-CMDB propellant, forming stable structures that mitigate the autocatalytic effect of free radicals, thereby reducing the burning rate.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
-
SHI Jun-hao, JIANG Tian-yu, ZHANG Wen-quan
Online:April 15, 2026 DOI: 10.11943/CJEM2026057
Abstract:Stereoisomerism plays a unique role in tuning the structures and performances of energetic molecules. Photochemical reactions feature mild conditions and precise configurational regulation thus bear important theoretical and practical significance for realizing stereoisomeric transformation of energetic molecules. (E)-Potassium 5,5'-azotetrazolate (E-PZT) was employed as the substrate in this work. Systematic condition screening was conducted to determine the optimal parameters for the photochemical synthesis of (Z)-potassium 5,5'-azotetrazolate (Z-PZT). The molecular structure of Z-PZT was fully characterized. The half-life of Z-PZT is measured to be 49 min at room-temperature. Theoretical calculations highly consistent with the experimental phenomena and results of the photoinduced isomerization reaction. The predicted excitation wavelengths and corresponding spectra obtained from theoretical calculations are highly consistent with the experimental phenomena and results of the photoinduced isomerization reaction. The successful preparation of the target product Z-PZT is further verified by these theoretical results. The isomerization energy barrier was calculated, and a rational photochemical reaction mechanism was accordingly proposed to elucidate the metastable characteristic of Z-PZT and the intrinsic origin of its relatively short half-life. Reliable experimental and theoretical support is provided by this work for the investigation of photoinduced isomerization regulation in energetic molecules.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
-
TANG Chang-wei, HE Dan, GE Zhong-xue, HE Jin-feng, ZHANG Ru-xin, WANG Xiao-ying
Online:April 20, 2026 DOI: 10.11943/CJEM2026029
Abstract:The inherent conflict between high energy density and low mechanical sensitivity represents a central challenge in the field of energetic materials. Although traditional nitramine compounds such as RDX, HMX, and CL-20 have significantly enhanced energy levels, they remain constrained by this trade-off. To explore new pathways for overcoming conventional performance limitations, researchers have proposed a strategy centered on rigid, planar fused-ring frameworks, leading to the development of nitrogen-rich fused-ring compounds. This review systematically outlines the evolution of this field, from the design of monocyclic systems (e.g., azoles, azines, and 1,2,5-oxadiazoles) to the integrated design of binary fused-ring systems. It highlights the conceptual design of representative molecules, key advances in synthetic methodologies—ranging from oxidative nitration to controlled rearrangement reactions—and the regulatory mechanisms of intermolecular interactions such as hydrogen-bonding networks and π-π stacking on material performance. This progression illustrates a paradigm shift from empirical trial-and-error to rational design and function-oriented customization. Finally, addressing the synthetic bottlenecks that constrain practical application, this review proposes that future breakthroughs require synergistic efforts across three dimensions: design, preparation, and application. This includes developing design methods that balance performance with synthetic feasibility, promoting synthetic technologies with improved safety profiles, and expanding the application scope of fused-ring energetic materials, thereby facilitating the transition from molecular design to practical implementation and providing a foundation for next-generation high-performance energetic materials.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
- 105
- 106
- 107
- 108
- 109
- 110
- 111
- 112
- 113
- 114
- 115
- 116
- 117
- 118
- 119
- 120
- 121
- 122
- 123
-
LIU Song-yuan, XIA Yu, CHENG Zhi-peng, LV Jia-lu, LEI Kang, LIANG Jian-hao, WU Xing-liang, XU Sen
Online:April 29, 2026 DOI: 10.11943/CJEM2026060
Abstract:To investigate the evolution of the mechanical sensitivity of the energetic oxidizer ammonium dinitramide (ADN) under varying moisture and temperature conditions, standard tests specified by the Federal Institute for Materials Research and Testing (BAM) were carried out in combination with the Langlie–D optimization method. The impact and friction sensitivities of ADN samples with moisture contents of 0, 5%, 10%, and 15% were quantitatively evaluated at 25, 50, and 75 ℃. The results show that both impact and friction sensitivities decrease significantly with increasing moisture content, although the variation is not simply linear and instead exhibits a distinct stagewise pattern. At 25 ℃, the limiting impact energy increased from 4 J for anhydrous ADN to 30 J at 5% moisture content and exceeded 50 J when the moisture content reached 10% or higher. Meanwhile, the minimum friction load increased progressively from 56 N and surpassed the upper measurement limit of the apparatus at 15% moisture content. Further analysis indicates that increasing temperature gradually weakens the desensitizing effect of moisture. At a given moisture content, both the limiting impact energy and the minimum friction load at 50 and 75 ℃ are generally lower than those at 25 ℃, while the corresponding impact and friction ignition-probability curves shift continuously toward lower stimulus levels. Among them, friction sensitivity shows a stronger dependence on temperature. These results suggest that the coupled effects of moisture content and temperature are reflected not only in single threshold values, but also in systematic shifts of the entire ignition-probability curve and the corresponding low-probability risk boundary.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
-
ZHANG Jing, DOU Jin-kang, TANG Chang-wei, GAO Min, TAN Bo-jun, LIU Ning
Online:May 07, 2026 DOI: 10.11943/CJEM2026039
Abstract:To investigate the regulatory mechanism of amino and nitro substituents on the thermal stability of TYX series heat-resistant explosives, two novel heat-resistant explosives based on the bis(triazolo)tetrazine backbone-fully amino-substituted 2,7-diaminobis([1,2,4]triazolo)[1,5-b:1',5'-e][1,2,4,5]tetrazine-5,10-diium-3,8-diide (TYX-1) and mono-nitro mono-amino substituted 2-amino-7-nitrobis([1,2,4]triazolo)[1,5-b:1',5'-e][1,2,4,5]tetrazine-5,10-diium-3,8-diide (TYX-3)-were selected in this study. Their thermal decomposition behaviors were systematically compared using differential scanning calorimetry (DSC) and thermal decomposition kinetic methods, while the decomposition processes were comprehensively analyzed by thermogravimetry-infrared-mass spectrometry (TG-FTIR-MS). The results show that the difference in substituents exerts a decisive influence on their thermal stability and decomposition pathways. TYX-1 exhibits a single high-temperature decomposition process with a peak temperature of 477.56 ℃ (at a heating rate of 20 ℃·min-1), and its decomposition mechanism conforms to the random two-dimensional nucleation growth model (A2), consistent with the layered stacking structure promoted by amino groups and the resulting controlled two-dimensional energy release pathway. In contrast, TYX-3 shows a significantly lower decomposition temperature and multi-step decomposition characteristics: the initial stage follows a two-dimensional diffusion model (D2), followed by a multi-reaction competitive stage, and finally transitions to a skeletal fracture process dominated by the one-dimensional chemical reaction model (F1). Gas product analysis shows that the main decomposition products of TYX-1 are N2, CO2, N2O, and HCN, while additional products including NO, HCNO, NH2, and H2O are detected for TYX-3, confirming that the nitro group, as a strong oxidizing moiety, induces an unconventional decomposition pathway and promotes the oxidative cleavage of the parent ring skeleton.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
-
HE Yan, WU Hao, ZHAI Lian-jie, CAI Rong-bin, XU Cheng, HU Jian-jian, HUANG Jun-rui, ZhAO Xue
2026,34(5):501-512, DOI: 10.11943/CJEM2026054
Abstract:To supplement the scarce basic solubility data of cyclotetramethylene tetranitramine (HMX) in dimethyl sulfoxide (DMSO)-alcohol mixed solvent systems and solve the technical bottleneck restricting the optimization of HMX crystallization process, the solubility of HMX in three binary mixed solvents of DMSO-methanol, DMSO-ethanol and DMSO-n-propanol was measured via the static method within the temperature range of 293.15-343.15 K. The experimental solubility data were correlated and fitted by the Apelblat model, Jouyban-Acree model and NRTL model, and the dissolution thermodynamic properties of HMX were further calculated and analyzed based on the NRTL model. The results show that the solubility of HMX in the three mixed solvent systems increases significantly with the rise of temperature and DMSO mole concentration. The Apelblat model exhibits the optimal correlation performance for the solubility data of the systems, with the average relative deviation (ARD) less than 5% and the root-mean-square deviation (RMSD) less than 0.11%, which is superior to the other two models. Thermodynamic analysis indicates that the dissolution process of HMX in all three DMSO-alcohol mixed solvent systems is endothermic, entropy-driven and spontaneous.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
-
CHEN Xin-ya, LI Li-jie, TAO Yu-ting, JIN Shao-hua, CHEN Kong, WANG Tong-bin, XU Zi-shuai
2026,34(5):513-520, DOI: 10.11943/CJEM2026006
Abstract:To investigate the effect of α-HMX content on the thermal safety of RDX produced via the acetic anhydride method, and thus addressing a gap in the existing research, five RDX samples (R1-R5) were prepared with α-HMX contents of 0, 9.50%, 12.41%, 17.00%, and 21.68%, respectively. The crystal form of HMX was identified using infrared spectroscopy and scanning electron microscopy. By employing differential scanning calorimetry (DSC) combined with the Ozawa method, Kissinger method, and modelling techniques, we systematically analyzed the thermal decomposition characteristics, kinetic patterns, and storage/transport safety parameters of the samples. Findings indicate that α-HMX reduces the activation energy and decomposition peak temperature of RDX. When the α-HMX content is ≤17.00%, compatibility remains satisfactory and thermal stability is maintained. However, when the α-HMX content reaches 21.68%, the apparent activation energy drastically decreases to 137.29 kJ·mol-1, compatibility deteriorates, and the self-accelerating decomposition temperature (SADT) drops to approximately 108 ℃, which markedly increases the risk of thermal detonation. Both the RDX and RDX/α-HMX systems follow a three-step sequential reaction model: ‘autocatalysis → N-stage → autocatalysis’. The Ozawa and Kissinger methods can provide initial parameter values for kinetic model fitting. For storage and transportation, there were no significant differences in the SADT among the groups at load capacities of 10, 20 kg and 50 kg. The kinetic model and critical safety parameters established herein provide essential technical support for production control and storage/transport safety in acetic anhydride-based RDX manufacturing, and confirm that the α-HMX content in the product must be maintained below 17.00%.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
-
ZHOU Fu-kang, YANG Xiao-yuan, SHANG Hai-lin, PAN Chuan-yu, LI Jin-he, LI Tao
2026,34(5):521-527, DOI: 10.11943/CJEM2026012
Abstract:To investigate the reaction-growth behavior of propellant and polymer-bonded explosive (PBX) under mass-inertial confinement following non-shock ignition, a thick-walled cylinder/mass-block experimental configuration was developed. Laser ignition was used to initiate the reaction. Multiple photonic Doppler velocimetry (PDV) probes were arranged to synchronously measure the radial expansion velocity of the cylinder and the axial velocity of the top surface of the mass block. High-speed imaging was also employed. Post-test residue recovery and examination were further conducted to comparatively analyze the reaction-evolution process. The results show that mass-inertial confinement intensifies the early-stage pressurization during reaction growth. Under identical confinement conditions, however, the type of energetic material governs the reaction-growth characteristics and reaction violence. In the propellant tests, the early-stage pressurization is dominated by mass-inertial confinement. The system response is characterized primarily by axial motion of the mass block, and no yielding deformation occurs in the thick-walled cylinder. The peak reaction pressure remains below 50 MPa, the reacted fraction is less than 1%, and nearly all of the propellant is recovered, indicating a burning event. In the PBX tests, the early-stage pressurization is jointly governed by mass-inertial confinement and structural confinement. The cylinder undergoes yielding and radial expansion, while the mass block exhibits axial motion accompanied by local upsetting deformation. The peak reaction pressure reaches 2 GPa, the reacted fraction exceeds 50%, and no explosive residue is recovered, indicating a violent explosive reaction.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
-
CHENG Bing, YE Fu, WANG Quan, CHENG Yang-fan, ZONG Qi, XU Ying, WANG Meng-xiang, LI Jun-hao
2026,34(5):528-538, DOI: 10.11943/CJEM2026001
Abstract:To improve the rock failure effects in open-pit deep hole blasting, a hole-inner layered column charge was designed. Firstly, small charge quantity blasting experiments under different charge forms were conducted to obtain the blasting failure process and final failure morphology of rock specimens. Then, the DEM-PBM (Discrete Element Method - Particle Blast Method) coupled simulation technique was used to numerically simulate the blasting process of various charges, further visually revealing the blasting failure characteristics of rock specimens. Finally, field tests were conducted to investigate the practical application effects of this charge form. The results showed that under the blasting action of hole-inner continuous column charge, only one blasting crack occurs in the 1/4 section at the top of the rock specimen, dividing it into two parts. The horizontal moving velocity of the rock fragment is 2.0 m·s-1, and a large rock fragment with a diameter of 9.0 cm is produced after blasting. Under the the blasting action of hole-inner layered column charge, multiple blasting cracks occur in the 1/4 section at the top of the rock specimen, dividing it into several fragments. The horizontal moving velocity of the rock fragment increase into 7.0 m·s-1, and no large fragments with a diameter of more than 5 cm is formed. Numerical simulations visualized the blasting process of rock specimens, and verified that the hole-inner layered column charge could eliminate large fragments with a size exceeding 5.0 cm. In practical open-pit deep hole bench blasting, the use of hole-inner layered column charge could significantly enhance the failure degree of rock mass at the top of bench, reducing the large rock fragment rate from 48.1% to 5.6%, which proved its engineering practicality in improving rock failure effects of deep hole blasting.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
- 105
- 106
- 107
- 108
- 109
- 110
- 111
- 112
- 113
- 114
- 115
- 116
- 117
- 118
- 119
- 120
- 121
- 122
- 123
- 124
- 125
- 126
- 127
- 128
- 129
- 130
- 131
- 132
- 133
- 134
- 135
- 136
- 137
- 138
- 139
- 140
- 141
- 142
- 143
- 144
- 145
- 146
- 147
- 148
- 149
- 150
- 151
- 152
- 153
- 154
- 155
- 156
- 157
- 158
- 159
- 160
- 161
- 162
- 163
- 164
- 165
- 166
- 167
- 168
- 169
- 170
- 171
- 172
- 173
- 174
- 175
- 176
- 177
-
WANG Ruo-xuan, WANG Shou-fei, TANG Jun-lan, ZOU Kai-jian, WANG Xiao-jun, ZHOU Lin-cheng, YE Zheng-fang
2026,34(5):539-550, DOI: 10.11943/CJEM2026019
Abstract:To address the treatment dilemma of HMX production wastewater characterized by complex components, high toxicity and poor biodegradability, a heterogeneous Fenton catalyst Fe2.5Mo/AC loaded with Fe/Mo bimetals was prepared via the impregnation-high temperature carbonization method using chitosan-modified activated carbon (AC) as the carrier. The amino functional groups of chitosan were adopted to anchor metal active sites and inhibit the agglomeration of active components; meanwhile, Mo was introduced to construct a Fe-Mo bimetallic synergistic system, which accelerated the Fe³⁺/Fe²⁺ redox cycle and improved the generation efficiency of ·OH. Results showed that a stable Fe-Mo-O/N composite structure was formed inside the catalyst with uniformly distributed active sites. Batch experimental results indicated that the catalytic system was applicable to a wide pH range of 2.09-7.15. The removal efficiencies of HMX and COD reached 83.1% and 44.5%, respectively, and the B/C ratio of wastewater was increased from 0.011 to 0.367, presenting an obvious improvement in biodegradability. The optimal reaction conditions were determined as catalyst dosage of 5 g·L-1, H2O2 concentration of 300 mM and reaction temperature of 25 ℃. Radical quenching experiments combined with electron paramagnetic resonance (EPR) tests verified that ·OH, 1O2 and ·O2- jointly participated in the degradation of HMX. After seven cycles of reuse, the HMX removal rate remained above 70% with extremely low metal leaching capacity. During 14 h continuous operation in the fixed bed reactor, the removal efficiencies of COD and HMX were stably maintained above 70% and 60%, respectively.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
- 72
- 73
- 74
- 75
- 76
- 77
- 78
- 79
- 80
- 81
- 82
- 83
- 84
- 85
- 86
- 87
- 88
- 89
- 90
- 91
- 92
- 93
- 94
- 95
- 96
- 97
- 98
- 99
- 100
- 101
- 102
- 103
- 104
- 105
- 106
- 107
- 108
- 109
- 110
- 111
- 112
-
PENG Yue, YUAN Xiao-xia, ZHANG Lei, XIE Ming-wei, MA Hong-liang, LI Fang
2026,34(5):551-563, DOI: 10.11943/CJEM2026061
Abstract:To improve and optimize the description of high-temperature and high-pressure behaviors of complex nanostructured carbon cluster products in explosives such as triaminotrinitrobenzene (TATB) and benzotrifuroxan (BTF), and effectively enhance the prediction accuracy of the detonation thermodynamic calculation program VPL for detonation parameters of the above explosives. Based on the physical model established by molecular dynamics research on carbon condensation, this study introduces a carbon phase fraction algorithm. Combined with the single-phase equations of state (EOS) of graphite and diamond, a novel equation of state named NDGP (Nano-Diamond-Graphite-Peng) is established to characterize diamond-graphite core-shell composite nanostructured carbon clusters. Meanwhile, by modifying the single-phase EOS of graphite, a new EOS denoted as NOCP (Nano-Onion-Carbon-Peng) is developed for describing onion-like nanocarbon clusters formed under high detonation temperature conditions. NDGP and NOCP are adopted to calculate the detonation velocity, detonation pressure, overdriven detonation Hugoniot relations and work capacity of detonation products for TATB (including TATB-based composite explosives) and BTF, respectively. By comparing the calculated results with the data obtained from two classic condensed carbon EOS (Fried and Cowan-Fickett models), it is verified that the two newly developed EOS for condensed carbon products can achieve more accurate prediction of C-J detonation parameters of TATB and BTF. Specifically, the prediction accuracy of TATB detonation velocity is generally increased by 1.5%-2.5%, and more accurate evaluation results can also be obtained for product work capacity and overdriven detonation Hugoniot relations. In addition, an EOS for characterizing the thermodynamic properties of low-density disordered carbon products is introduced. The detonation velocity-density correlation of the typical primary explosive lead trinitroresorcinate (LTNR) is systematically calculated. Compared with the Fried and Cowan-Fickett EOS, the overall prediction accuracy is improved by 3%-7%.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
-
SANG Sheng-jie, TONG Zhi-ke, MIAO Yu-xin, SHI Jun-li, ZHANG Huan-ling, LI Shuo, LI Xiang-zhi, BI Fu-qiang, CAO Duan-Lin, ZHAO Lin-xiu
2026,34(5):564-571, DOI: 10.11943/CJEM2026051
Abstract:Tubular channel reactors possess superior advantages of efficient mass and heat transfer, which lays a solid foundation for the industrial transformation of mixing-controlled strongly exothermic reactions from traditional batch processes to continuous-flow processes. In this work, the synthesis of 3-amino-4-aminoximinofurazan (AAOF) was taken as the research subject. First, reaction calorimetry experiments were carried out to acquire the fundamental heat release data during the reaction process. Combined with material balance and heat balance calculations, the characteristic parameters of the exothermic model inside the tubular channel reactor were determined, and a coupled heat transfer-exothermic model was subsequently established. A numerical solution method was employed to simulate the jacket heat transfer capacity, heat transfer rate, and heat exchange efficiency of the heat transfer medium. On the basis of the simulation results, the thermal safety risks existing in the continuous-flow reaction process were analyzed, and a targeted heat exchange control scheme was proposed accordingly.The results reveal that under the fixed structural parameters of the tubular channel reactor (inner diameter of 0.01 m, tube length of 5 m) and AAOF production capacity of 2 kg·h-1, when heat conduction oil is used as the heat exchange medium in the cocurrent flow mode, its mass flow rate exerts a remarkable influence on process thermal safety. When the mass flow rate is lower than 0.1 kg·h-1, the outlet temperature of the tubular reactor exceeds 120 ℃, approaching the initial decomposition temperature of AAOF reaction solution (121.7 ℃), which is liable to induce heat accumulation and further reaction runaway. The optimal heat removal performance is achieved at a mass flow rate range of 2-3.5 kg·h-1. When the mass flow rate exceeds 4.5 kg·h-1, the system temperature drops below 100 ℃ and cannot satisfy the required process temperature conditions. The optimal mass flow rate range of the heat transfer medium is finally determined to be 2-3.5 kg·h-1. This study provides basic fundamental data and parametric guidance for the process safety design and stable long-term operation of AAOF preparation in tubular channel reactors.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
-
LIU Shi-shuai, MA Li, GUO Xiao-wei, JIANG Xia-bing, ZHAO Yuan-yu
2026,34(5):572-580, DOI: 10.11943/CJEM2026023
Abstract:To address the challenges of complex components, difficult identification, and low intelligence of traditional detection methods for mixed explosives, two energetic material mixtures of m-dinitrobenzene/potassium nitrate and p-nitroaniline/ammonium nitrate were selected as research objects. A sequential detection strategy combining infrared preliminary screening and Raman confirmation was adopted. Combined with convolutional neural network (CNN)-based deep learning intelligent spectral image processing and recognition method, the spectral response characteristics of the samples in powder and flake forms were investigated. Meanwhile, the effects of component content and physical morphology on detection results were explored. The results indicate that for powdered energetic material samples, infrared spectroscopy can preliminarily identify the presence of m-dinitrobenzene and p-nitroaniline via characteristic peaks at specific wavenumbers, whereas it is difficult to independently distinguish inorganic oxidants such as potassium nitrate and ammonium nitrate. Raman spectroscopy can effectively characterize the nitrobenzene functional group structures of both powdered and flake samples. It can not only realize the qualitative identification of organic energetic components, but also detect characteristic signals unresponsive to infrared spectroscopy, thereby achieving accurate full-component identification of mixed explosives. Although instrumental parameters, excitation wavelength and sample morphology cause spectral peak shift and intensity fluctuation, the positions of core characteristic peaks and overall spectral profiles maintain favorable stability, which can provide a reliable spectral basis for the classification and identification of mixtures. The average recognition accuracy of the deep learning-based intelligent recognition model reaches 96.54% and 96.29% for mid-infrared and Raman spectral samples, respectively, with the average recognition time of a single sample being 0.044 s and 0.042 s.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
-
ZHANG Shu-sheng, LIU Jian, WANG Tao, ZHANG Chao-yang, LIU Yu
2026,34(5):589-591, DOI: 10.11943/CJEM2026066
Abstract:Energy level is the fundamental property of energetic compounds and is one of the core criterion for determining whether an energetic compound has application potential. Experimental determination of detonation parameters is the basic method to evaluate energy level. However, due to the large dosage required and the poor reproducibility of the result, it is impossible to directly provide the theoretical maximum energy, which fails to meet the needs of new compound development. Non-experimental methods, including detonation equations of state, semi-empirical formulas and machine learning models, can predict detonation parameters via basic physicochemical properties or even only molecular structures. These methods become effective approaches for designing and screening new energetic compounds. This study systematically reviews these non-experimental evaluation methods. It summarizes and analyzes their basic principles, applicable conditions, advantages and disadvantages. Equations of state are theoretically rigorous and reliable but need precise physicochemical properties as input. Semi-empirical formulas are the most convenient to apply but lack sufficient accuracy and still require physicochemical properties as input. Machine learning models can predict detonation parameters only by molecular structures but have poor generalization performance. Overall, all existing methods are established based on available experimental data. Their weak data foundation limits extrapolation capability for unknown compounds, failing to meet the requirements for the design and screening of novel energetic compounds. Herein, we propose physics-mechanism-constrained hybrid models and dimensional expansion of energy evaluation parameters, to overcome the experimental data dependence of existing methods and improve their prediction accuracy and generalizability.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
-
PENG Guo-bin, ZHAO Peng-cheng, DUAN Jia-he, SONG Hui, NAN Ce
2026,34(5):592-609, DOI: 10.11943/CJEM2025259
Abstract:Gunshot residue (GSR) is a trace particle formed during the firing of a bullets. As an important research subject in forensic science, it plays a key role in the investigation of gun-related cases. Currently, conventional GSR detection mainly relies on large laboratory instruments. However, due to the complexity of sample pretreatment and the longthy submission process, it is difficult to provide analysis results quickly, thereby affecting the decision-making efficiency of on-site investigation work. In recent years, GSR on-site rapid detection technology has received widespread attention due to its simple operation, low cost, and portability. This type of technology can be directly implemented at the crime scene without relying on large precison instruments , and can quickly output detection results. It is suitable for the preliminary screening of GSR and can also be used as the final confirmation detection method, and has become a research hotspot in this field. Therefore, a systematic review of the research progress on-site rapid detection technology for GSR is conducted, focusing on introducing five categories of methods: colorimetric methods, spectroscopic methods, mass spectrometry methods, electrochemical methods, and fluorescent labeling methods. Their advantages and limitations are thoroughly analyzed, and they are compared with the practical applications of laboratory detection technology. Finally, future research development directions are proposed, aiming to provide theoretical basis and methodological references for on-site technicians in actual detection work.
- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
Vol, 34, No.5, 2026
>Research Articles




































































































































































































































































































































































































































































































































































































































>Reviews



































































-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
Propellant
2021-2023 Collection
-
![]()
-
![]()
Gun Propellant
2021-2023 Collection
-
![]()
Safety and damage study
2021-2023 Collection
-
![]()
Initiator and Pyrotechnics
2021-2023 Collection
-
![]()
Preparation and Property
2021-2023 Collection
-
![]()
Crystal and microscopic analysis
2020-2022 发表
-
![]()
Chemical Propellant
2021-2022 Collection
-
![]()
-
![]()
-
![]()
-
![]()
Eco-friendly technology
2021-2022 Collection
-
![]()
Initiating explosive device technology
2021-2022 Collection
-
![]()
Damage and ignition
2021-2022 Collection
-
![]()
Thermal decomposition,safety performance and evaluation
2021-2022 Collection
-
![]()
Preparation and performance—Characterization of molding materials
2021-2022 Collection
-
![]()
Preparation and performance—Characterization of synthesis
2021-2022 Collection
-
![]()
Preparation and performance—Study on synthesis and performance
2021-2022 Collection
-
![]()
Explosion and damage
2021-2022 Collection
-
![]()
Detonation physics of energetic materials
2021-2022 Collection
-
![]()
High efficiency destruction technology
2021-2022 Collection
-
![]()
Propulsion and projection—Propulsion Materials structure and activity relationship
2021-2022 Collection
-
![]()
Propulsion and projection—Preparation and performance about propulsion materials
2021-2022 Collection
-
![]()
Calculation and simulation—Material structure and response
2021-2022 Collection
-
![]()
Calculation and simulation—Structural evolution of materials
2021-2022 Collection
-
![]()
Calculation and simulation—Material performance prediction
2021-2022 Collection
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
-
![]()
Damage and Ignition
2020
-
![]()
Detonation Physics
2020
-
 Special Issue
Special Issue










































 Virtual Special Issue
Virtual Special Issue









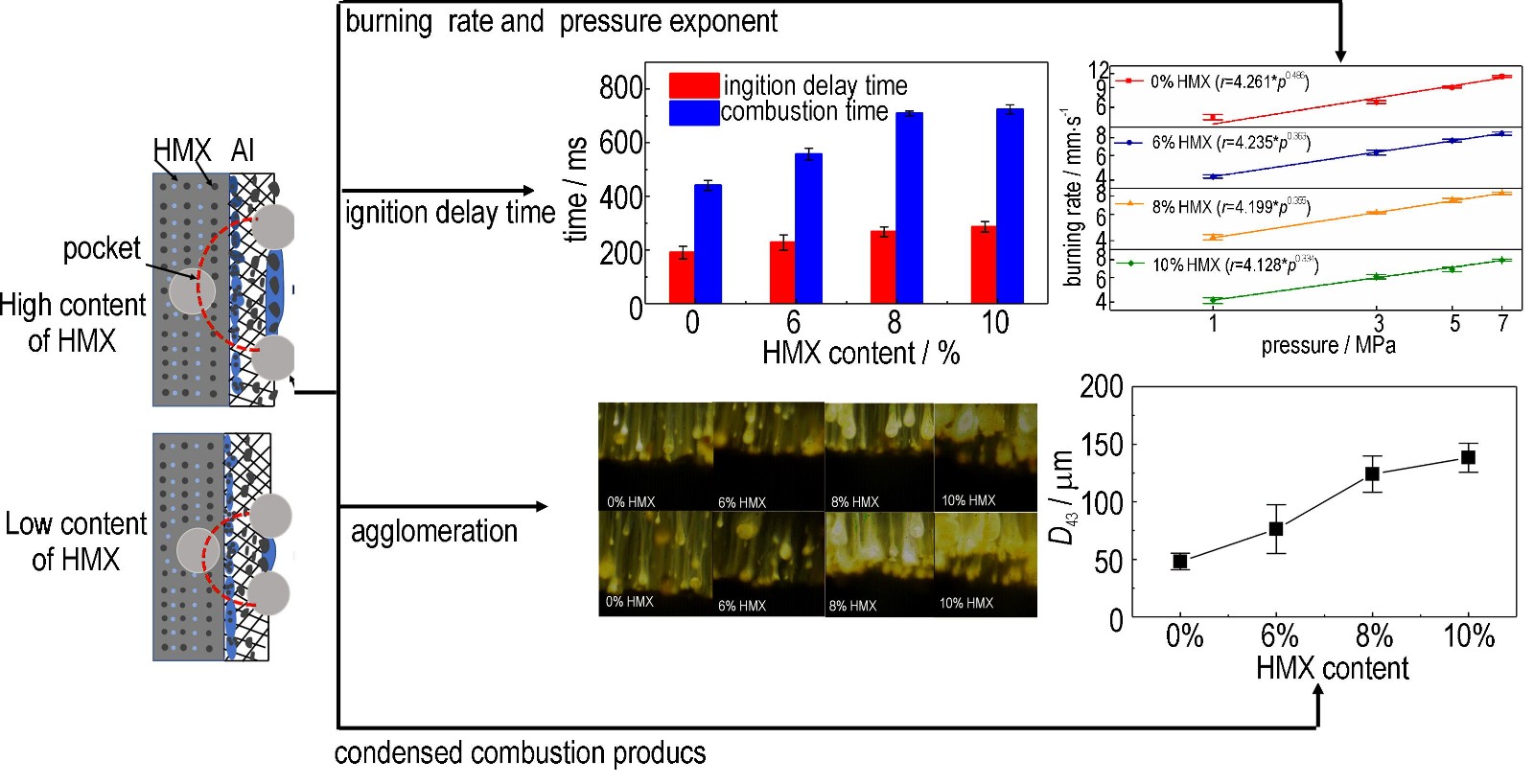



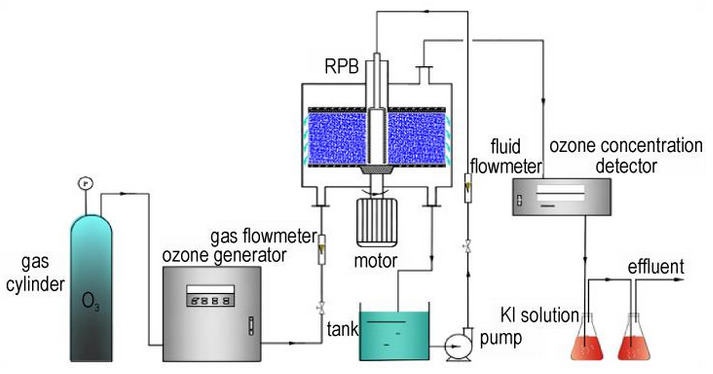
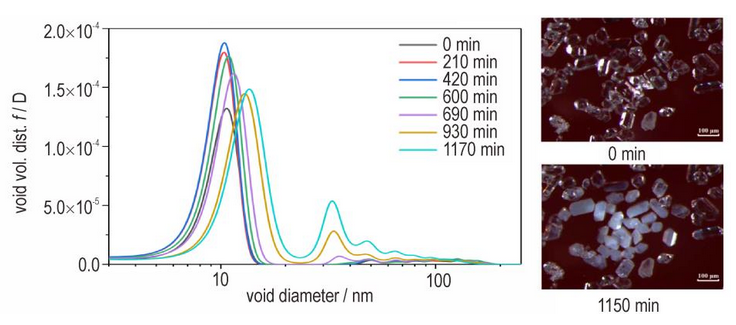
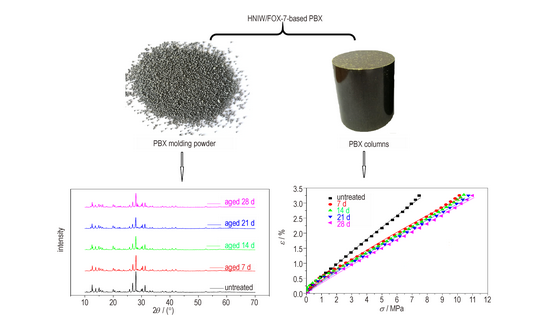
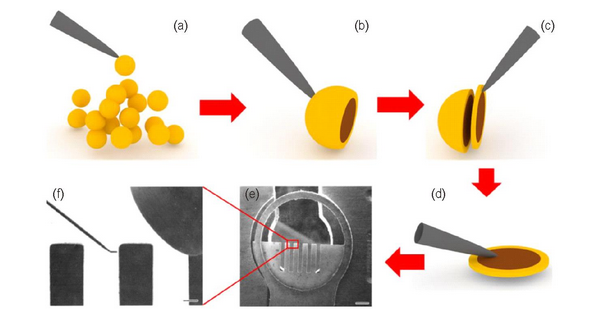
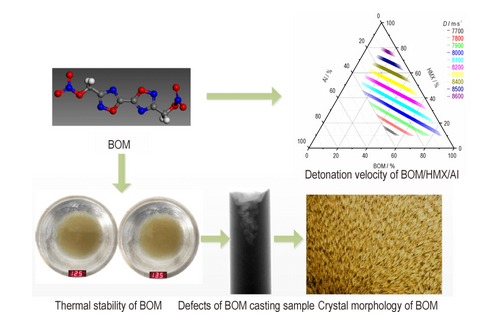
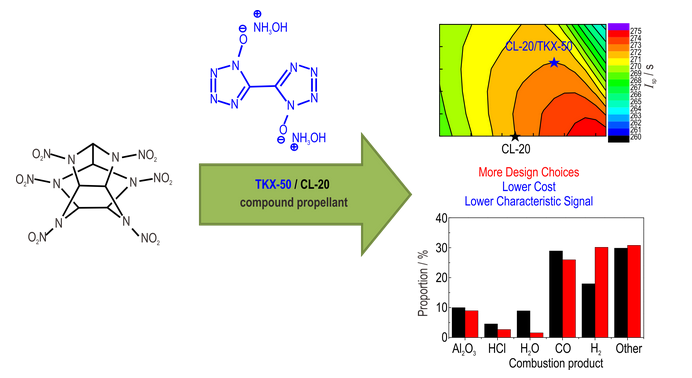
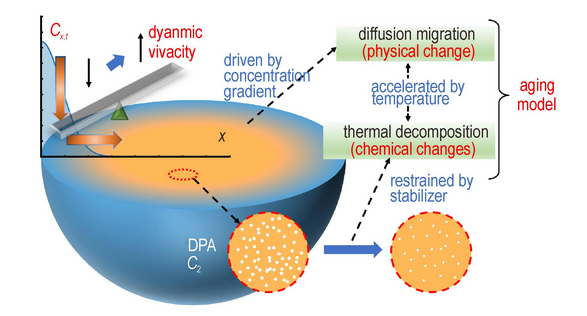
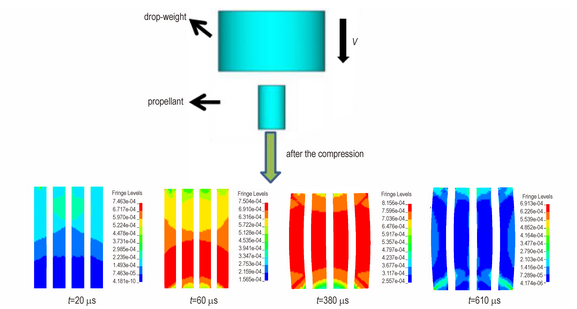
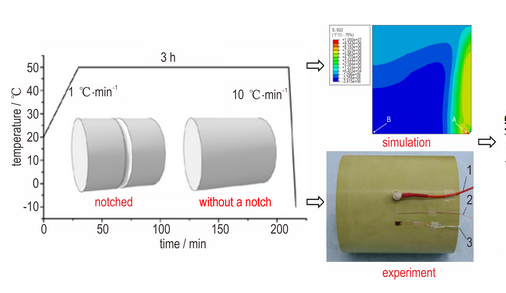
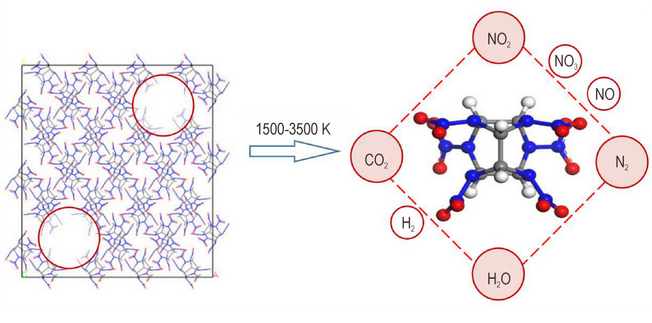














CJCR

