




2. 嘉兴学院生物与化学工程学院, 浙江 嘉兴 314001
2. College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, China
太安(PETN, 季戊四醇四硝酸酯,

|
图 1 PETN的分子结构和原胞结构 Fig.1 Molecular structure and primitive cell structure of PETN |
感度即安全性,是高能物质最重要性能之一。先前的微观理论判据主要是通过量子化学计算求得的, 用于高能分子较多[15]。对于成千上万原子的超晶胞或复合材料, 近年来已有关于HMX/AP[16, 17]、HMX基PBX[18, 19]、RDX完美和缺陷晶体及RDX基PBX[20, 21]感度判别的工作发表。我们所建议的引发键最大键长(
力学性能也是高(含)能材料最重要的性能之一。过去的十年, 本课题组已开辟了高能复合材料(PBXs、发射药和推进剂)弹性力学性能的预测研究[22-30], 主要都是基于静态力学分析。而收集MD平衡后的原子运动全轨迹加以分析的波动法[31], 虽然耗机时较多, 但对力学性能的预测相对要准确可靠, 故本研究试以波动法求得PETN晶体的力学性能。
在物质结构-性能微观理论研究中, 经典MD模拟较量子力学(QM)计算有一个优点, 就是能求得庞大复杂体系结构、性能随温度递变的统计平均规律。因为研究易爆燃物质的感度和力学性能及其温度效应极为重要, 故而本实验以PETN晶体为研究对象, 选择构建其(4×3×4)超晶胞, 并选择其较为钝感的(100)面[3, 13, 32]加以切割分面, 将所得这两种模型, 在5种不同温度(195, 245, 295, 345,395 K)和NPT系综下, 进行MD模拟研究, 重点探讨结构、能量和力学性能及其温度效应, 寻求和核准微观感度理论判据。
2 模型搭建和模拟细节 2.1 力场选择和模型搭建本研究之所以选择COMPASS力场[33], 是因为该力场能较好地适用于凝聚态, 包括硝酸酯及其相关物[34-36]。以PETN的X射线衍射晶体数据[37-38]为依据, 构建PETN(4×3×4)超晶胞并置于具周期性边界条件的周期箱中; 每个周期箱中含96个PETN分子, 共2784个原子, 即为PETN晶体模型。沿PETN(4×3×4)超晶胞(100)晶面方向进行“切割”, 并使Z轴平行晶轴

|
图 2 PETN和PETN(100)的初始模型 Fig.2 Primary model of PETN and PETN (100) |
将搭建好的PETN和PETN(100)两种晶体模型分别在COMPASS[33]力场、NPT系综下进行MD周期性模拟研究。模拟过程中温度和压力的控制采用Anderson [39]和Parrinello[40]方法, 范德华(vdW)和静电作用(Coulomb)分别用Atom-Based[41]和Ewald[42]方法, 截断半径取9.5×10

|
图 3 PETN和PETN(100)晶体的平衡结构 Fig.3 Equilibrium cells of PETN and PETN(100) |
表 1给出PETN晶体295 K下NPT-MD模拟所得晶胞参数和密度。为方便比较, 表中还给出了实验值[38]和Sorescu等的理论计算结果[1, 10, 43]。由表 1可见, 本工作模拟所得晶胞参数与前人的实验和计算值吻合较好, 晶胞参数相差很小或相等; 与实验密度相对误差也只有0.6%。由此进一步表明COMPASS力场对PETN晶体有较好的适用性, 也表明本研究的MD模拟确已达到平衡, 所得平衡结构可信。
| 表 1 PETN晶胞参数和密度的理论计算值与实验值比较 Tab.1 Comparison of calculated and experimental results of PETN lattice parameters and density |
MD模拟能提供各种体系在广义平衡结构下的键长统计分布, 包括在不同温度下的键长分布, 这是MD很有意义的优势所在。PETN的引发键一般认为是分子中的O—

|
图 4 PETN和PETN(100)晶体中O— |

|
图 5 PETN和PETN(100)晶体中O— |
|
表 2 PETN和PETN(100)晶体中O— |
由图 4可见, PETN和PETN(100)的引发键键长分布均呈近似对称的高斯型分布, 最可几键长和平均键长较为接近。PETN晶体引发键键长在1.350~1.480Å范围的占97%, PETN(100)在1.345~1.485Å范围的占98%。由表 2可见, 它们在295 K下的
定义硝酸酯类体系中引发键(O—
表 3和表 4给出PETN和PETN(100)晶体在不同温度下的
|
表 3 不同温度下PETN晶体中的O— |
|
表 4 不同温度下PETN(100)中的O—N双原子作用能和相关分量
Tab.4 Interaction energy |

|
图 6 PETN和PETN(100)晶体中引发键连双原子作用能随温度的变化
Fig.6 Interaction energy ( |
内聚能密度(CED)是单位体积1 mol凝聚态克服分子间作用力变为气态时所需能量。在MD模拟中CED是vdW力与静电力之和, 即分子的非键力。基于MD模拟轨迹所得不同温度下PETN晶体和PETN(100)的内聚能密度及其分量。见表 5。
| 表 5 不同温度下PETN和PETN(100)晶体的内聚能密度和相关分量 Tab.5 Cohesive energy density CED and its components of PETN and PETN(100) |
由表 5可见, 随温度升高, PETN和PETN(100)的CED、vdW力和静电力均单调递减, 表明它们由晶态变为气态时所需能量变小, 这与温度升高感度增大的实验事实相一致。由此表明, 在一定条件下, PETN晶体的CED也可用于热感度相对大小的理论判据; 且在每个温度下PETN和PETN(100)的CED及其分量数值都惊人地一致, 表明CED的统计平均值不受MD模型的影响。
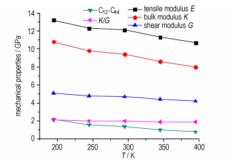
3.5 力学性能比较弹性模量是评价材料刚性的指标, 是材料抵抗弹性形变能力的度量[44]。而弹性性能和塑性性能是相关联的[45], 通常表示阻止塑性形变能力的硬度和拉伸强度与剪切模量呈正比; 断裂强度与本体模量呈正比; 本体模量与剪切模量的比值(
| 表 6 不同温度下PETN和PETN(100)晶体的力学性能 Tab.6 Mechanical properties at different temperatures for PETN and PETN (100) crystal |

|
图 7 PETN(100)晶体的力学性能随温度的变化 Fig.7 Mechanical properties versus temperature for PETN(100) |
根据广义虎克定律, 反映应力应变关系的弹性系数应有36个; 因
通过PETN和PETN(100)晶体两种模型在5个温度下的NPT-MD模拟, 得出如下主要结论:
(1) 模拟所得PETN晶胞参数和密度与实验值接近, 表明COMPASS力场适用、所得平衡结构可信。
(2) 模拟求得PETN晶体的引发键(O—
(3) PETN晶体的引发键连双原子作用能(
(4) 由波动法求得PETN晶体的力学性能。弹性系数和弹性模量随温度升高而减小, 表明各向同性递增, 刚性降低。
(5) 切割分面研究PETN晶体, 能获得较确定的结构、相互作用能和力学性能及其温度效应。
| [1] |
Sorescu D C, Rice B M, Thompson D L. Theoretical studies of the hydrostatic compression of RDX, HMX, HNIW, and PETN crystals[J].
The Journal of Physical Chemistry B, 1999, 103(32): 6783-6790. DOI:10.1021/jp991202o |
| [2] |
Zoui A, Sekkal W. Molecular dynamics study of mechanical and thermodynamic properties of pentaerythritol tetranitrate[J].
Solid State Cmmunications, 2001, 118(7): 345-350. DOI:10.1016/S0038-1098(01)00136-3 |
| [3] |
Wu C J, Ree F H, Yoo C S. A quantum mechanical molecular dynamics study of binary collisions of pentaerythritol tetranitrate (PETN): Its correlation to shock sensitivity[J].
Propellants, Explosives, Pyrotechnics, 2004, 29(5): 296-303. DOI:10.1002/(ISSN)1521-4087 |
| [4] |
Wu C J, Manaa M R, Fried L E. A molecular dynamics study of chemical reactions of solid pentaerythritol tetranitrate at extreme conditions[R]. Lawrence Livermore National Laboratory(LLNL), Livermore, CA, 2006.
|
| [5] |
Wu C J, Manaa M R, Fried L E. Tight binding molecular dynamic simulation of PETN decomposition at an extreme condition[J].
Mater Res Soc Symp Proc, 2007, 987: 139-144. |
| [6] |
Lin P H, Khare R, Weeks B L, et al. Molecular modeling of diffusion on a crystalline pentaerythritol tetranitrate surface[J].
Applied Physics Letters, 2007, 91(10): 104107 DOI:10.1063/1.2783206 |
| [7] |
Jian Wang, Ted Golfinopoulos, Richard H. Gee, et al. Diffusion on (110) surface of molecular crystal pentaerythritol tetranitrate[J].
Appl Phys Lett, 2007, 90(10): 101906 DOI:10.1063/1.2709955 |
| [8] |
Landerville A C, Oleynik I I, White C T. Reactive molecular dynamics of detonating PETN[C]∥AIP Conference Proceedings. 2009, 1195: 813.
|
| [9] |
Landerville A C, Oleynik I I, White C T. Reactive molecular dynamics of hypervelocity collisions of PETN molecules[J].
The Journal of Physical Chemistry A, 2009, 113(44): 12094-12104. DOI:10.1021/jp905969y |
| [10] |
Maiti A, Gee R H. Modeling growth, surface kinetics, and morphology evolution in PETN[J].
Propellants, Explosives, Pyrotechnics, 2009, 34(6): 489-497. |
| [11] |
Burnett A D, Kendrick J, Cunningham J E, et al. Calculation and measurement of terahertz active normal modes in crystalline PETN[J].
Chem Phys Chem, 2010, 11(2): 368-378. DOI:10.1002/cphc.v11:2 |
| [12] |
Pereverzev A, Sewell T D. Effect of vacancy defects on the terahertz spectrum of crystalline pentaerythritol tetranitrate[C]∥AIP Conference Proceedings, 2012, 1426: 1187.
|
| [13] |
Shan T R, Wixom R R, Mattsson A E, et al. Atomistic simulation of orientation dependence in shock-induced initiation of pentaerythritol tetranitrate[J].
The Journal of Physical Chemistry B, 2013, 117(3): 928-936. DOI:10.1021/jp310473h |
| [14] |
Yanilkin A, Sergeev O. Molecular dynamics simulation of the burning front propagation in PETN[J].
Bulletin of the American Physical Society, 2013, 58(7) |
| [15] |
肖鹤鸣, 朱卫华, 肖继军, 等. 含能材料感度判别理论研究——从分子, 晶体到复合材料[J].
含能材料, 2012, 20(5): 514-527. XIAO He-ming, ZHU Wei-hua, XIAO Ji-jun, et al. Theoretical studies on sensitivity criterion of energetic materials—from molecules, crystals, to composite materials[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(5): 514-527. |
| [16] |
朱伟, 肖继军, 郑剑, 等. 高能混合物的感度理论判别—不同配比和不同温度AP/HMX的MD研究[J].
化学学报, 2008, 66(23): 2592-2596. ZHU Wei, XIAO Ji-jun, ZHENG Jian, et al. A Theoretical criterion for sensitivity of energetic composites—molecular dynamics studies on AP/HMX systems at various concentrations and temperatures[J]. Acta Chimica Sinica, 2008, 66(23): 2592-2596. DOI:10.3321/j.issn:0567-7351.2008.23.004 |
| [17] |
Zhu W, Wang X, Xiao J J, et al. Molecular dynamics simulations of AP/HMX composite with a modified force field[J].
Journal of Hazardous Materials, 2009, 167(1): 810-816. |
| [18] |
Xiao J J, Wang W R, Chen J, et al. Study on the relations of sensitivity with energy properties for HMX and HMX-based PBXs by molecular dynamics simulation[J].
Physica B, 2012, 407(17): 3504-3509. DOI:10.1016/j.physb.2012.05.010 |
| [19] |
Xiao J J, Wang W R, Chen J, et al. Sensitivity and mechanical properties of HMX and HMX-based PBXs with molecular dynamics simulation[J].
Computational and Theoretical Chemistry, 2012, 999: 21-27. DOI:10.1016/j.comptc.2012.08.006 |
| [20] |
Xiao J J, Zhao L, Zhu W, et al. Molecular dynamics study on the relationships of modeling, structural and energy properties with sensitivity for RDX-based PBXs[J].
Science China Chemistry, 2012, 55(12): 2587-2594. DOI:10.1007/s11426-012-4797-1 |
| [21] |
Xiao J J, Li S Y., Chen J., et al. Molecular dynamics study on correlation between structure and sensitivity of defective RDX crystals and their PBXs[J].
J Mol Model, 2013, 19(2): 803-809. DOI:10.1007/s00894-012-1607-9 |
| [22] |
肖继军, 方国勇, 姬广富, 等. HMX基高聚物粘结炸药(PBX)结合能和力学性能的模拟研究[J].
科学通报, 2004, 49(24): 2520-2524. XIAO Ji-jun, FANG Guo-yong, JI Guang-fu, et al. Simulation investigations in the binding energy and mechanical properties of HMX-based polymer-bonded explosives[J]. Chinese Science Bulletin, 2004, 49(24): 2520-2524. DOI:10.3321/j.issn:0023-074X.2004.24.004 |
| [23] |
肖继军, 马秀芳, 黄玉成, 等. TATB/氟聚物PBX力学性能的分子动力学模拟[J].
含能材料, 2004, 12: 488-492. XIAO JI-jun, MA Xiu-fang, HUANG Yu-cheng, et al. Molecular dynamic simulation of mechanical properties of TATB/Fluorine-polymers PBX[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12: 488-492. |
| [24] |
马秀芳, 肖继军, 黄辉, 等. 分子动力学模拟浓度和温度对TATB/PCTFE PBX力学性能的影响[J].
化学学报, 2005, 63(22): 2037-2041. MA Xiu-fang, XIAO JI-jun, HUANG Hui, et al. Effects of concentration and temperature on mechanical properties of TATB/PCTFE PBX by molecular dynamics simulation[J]. Acta Chimica Sinica, 2005, 63(22): 2037-2041. DOI:10.3321/j.issn:0567-7351.2005.22.004 |
| [25] |
Xu X J, Xiao H M, Xiao J J, et al. Molecular dynamics simulation for pure-CL-20 and ε-CL-20-based PBXs[J].
J Phys Chem B, 2006, 110(14): 7203-7207. DOI:10.1021/jp060077v |
| [26] |
Xiao J J, Ma X F, Zhu W, et al. Molecular dynamics simulations of polymer-bonded explosives(PBXs): modeling, mechanical properties and their dependence on temperatures and concentrations of binders[J].
Propellants, Explosives, Pyrotechnics, 2007, 32(5): 355-359. DOI:10.1002/(ISSN)1521-4087 |
| [27] |
Qiu L, Zhu W H, Xiao J J, et al. Theoretical studies of solid bicyclo-HMX: effects of hydrostatic pressure and temperature[J].
Journal of Physical Chemistry B, 2008, 112(13): 3882-3893. DOI:10.1021/jp070863f |
| [28] |
Ma X F, Zhu W H, Xiao J J, et al. Molecular dynamics study of the structure and performance of simple and double bases propellants[J].
Journal of Hazardous Materials, 2008, 156(1): 201-207. |
| [29] |
Xiao J J, Huang H, Li J S, et al. A molecular dynamics study of interface interactions and mechanical properties of HMX-based PBXs with PEG and HTPB[J].
Journal of Molecular Structure: Theochem, 2008, 851(1): 242-248. |
| [30] |
Xu X J, Xiao J J, Huang H, et al. Molecular dynamics simulations on structures and properties of ε-CL-20(001)/F2314 PBX[J].
Journal of Hazardous Materials, 2010, 175: 423-428. DOI:10.1016/j.jhazmat.2009.10.023 |
| [31] |
Parrinello M, Rahman A. Strain fluctuations and elastic constants[J].
J Chem Phys, 1982, 76: 2662-2666. DOI:10.1063/1.443248 |
| [32] |
Dick J J. Shock initiation of detonation in-irradiated pentaerythritol tetranitrate[J].
J Appl Phys, 1982, 53(9): 6161-6167. DOI:10.1063/1.331527 |
| [33] |
Sun H. COMPASS: An ab initio forcefield optimized for condensed-phase application-overview with details on alkane and benzene compounds[J].
J Phys Chem B, 1998, 102(38): 7338-7364. DOI:10.1021/jp980939v |
| [34] |
Bunte S W, Sun H. Molecular modeling of energetic materials: the parameterization and validation of nitrate esters in the COMPASS forcefield[J].
Phys. Chem B, 2000, 104(11): 2477-2489. DOI:10.1021/jp991786u |
| [35] |
肖继军, 王艳群, 詹炜, 等. PETN基PBX结合能和力学性能的理论研究[J].
分子科学学报, 2006, 22(4): 219-225. XIAO JI-jun, WANG Yan-qun, ZHAN Wei, et al. Theoretical study on binding energies and mechanical properties of PETN-based PBX[J]. Journal of Molecular Science, 2006, 22(4): 219-225. |
| [36] |
王艳群, 肖继军. PETN为基的高聚物粘结炸药(PBX)力学性能的MD模拟[J].
长江大学学报(自科版)理工卷, 2007, 4(2): 44-46. WANG Yan-qun, XIAO JI-jun. MD simulation study on mechanical properties of PETN-based PBX[J]. Journal of Yangtze University (Nat Sci Edit) Sci Eng V, 2007, 4(2): 44-46. |
| [37] |
Trotter J. Bond length and angles in pentaerythritol tetranitrate[J].
Acta Cryst, 1963, 16: 698-699. DOI:10.1107/S0365110X6300178X |
| [38] |
Cady H H, Larson A C. Pentaerythritol tetranitrate Ⅱ: its crystal structure and transformation to PETN I; an algorithm for refinement of crystal structures with poor data[J].
Acta Cryst B, 1973, 31: 1864-1869. |
| [39] |
Andersen H C. Molecular dynamics simulations at constant pressure and/or temperature[J].
Chem Phys, 1980, 72(4): 2384-2393. |
| [40] |
Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method[J].
Appl Phys, 1981, 52(12): 7182-7190. DOI:10.1063/1.328693 |
| [41] |
Ewald P P. Evaluation of optical and electrostatic lattice potentials[J].
Ann Phys, 1921, 64: 253-287. |
| [42] |
Allen M P, Tildesley D J.
Computer Simulation of Liquids[M]. Oxford: Oxford University Press, 1989 |
| [43] |
Velizhanin K A., Kilina S, Sewell T D, et al. First-Principles-Based calculations of vibrational normal modes in polyatomic materials with translational symmetry: application to PETN molecular crystal[J].
The Journal of Physical Chemistry B, 2008, 112(42): 13252-13257. DOI:10.1021/jp804980a |
| [44] |
Weiner J H.
Statistical Mechanics of Elasticity[M]. NewYork: John Wiley, 1983 |
| [45] |
Pugh S F. Relation between the elastic moduli and the plastic properties of polycrystalline pure metals[J].
Phil Mag, 1954, 45(367): 823-843. DOI:10.1080/14786440808520496 |
| [46] |
Pettifor D G. Theoretical predictions of structure and related properties of intermetallics[J].
Mater Sci Technol, 1992, 8(4): 345-349. DOI:10.1179/mst.1992.8.4.345 |
Two different PETN crystalline models were simulated at five different temperatures by molecular dynamics (MD) simulation using COMPASS force field in the isothermal-isobaric (NPT) ensemble. The trigger bond length, the interaction energy between two atoms of the trigger bond and the cohesive energy density as well as the mechanical properties of the PETN crystal were presented and analyzed.