




混合炸药是由两种或两种以上的含能组分和粘结剂、增塑剂、钝感剂等添加剂按适当比例混合而成的, 其各组分性能优势可以互补, 适应多种使用要求, 因此是含能材料在武器中应用的主要形式[1]。混合炸药的凝聚态结构、相容性、安全性、力学和爆轰性能等在本质上均与分子间相互作用有关; 随着新型高能化合物、纳米材料以及功能高聚物的应用和炸药改性研究的深入[2], 分子间作用的研究显得至关重要。目前, 含能材料体系中分子间相互作用的实验研究主要以宏观表征方法为主, 如差示扫描量热法(DSC)[3, 10]、DSC-TG/DTG联用[4]、高压DSC(PDSC)-TG/DTG联用[5-6]、DSC/TG-MS联用[7]、量气法[4, 8-10]等热分析方法, 以及动态接触角测量[11-12]、界面张力测试[11-12]、动态热机械测试[13]等材料测试方法, 直接以组分混合后体系性能的变化来推测组分之间的相互作用, 并未涉及到分子间作用本质。此外, 扫描电镜(SEM)[14-15]、X射线光电子能谱(XPS)[14-17, 18]、傅里叶红外光谱(FTIR)[18]、显微红外光谱(MIR)[15-17]、X射线衍射(XRD)[19]等先进表征技术, 也可用来研究含能材料体系分子间相互作用机理, 这些实验结果为理论研究提供了可靠的依据。然而, 对于含能体系来说, 实验研究成本昂贵, 而且始终存在安全问题; 在技术层面, 对于分子间弱相互作用的电子结构层次的分析等还不够详尽; 因此进行理论研究既是对实验研究的补充也是一种安全有效的研究手段。近年来, 计算机模拟技术如量子化学(QC)、分子力学(MM)、分子动力学(MD)等[20-28]在含能材料领域得到了广泛的应用和飞速的发展。大量实验事实表明, 混合炸药体系通常是一种复杂的共混物体系, 其中存在有单质炸药的聚集体、不同炸药分子间、炸药与其他组分间、添加组分间以及组分界面间等不同情形的相互作用, 每一种情形都对整个混合体系的结构性能有所影响, 各层次计算机模拟方法的应用为深入分析解决相关问题提供了有效工具。本文在总结这些计算机模拟技术特点的基础上, 重点论述了其在混合炸药分子间相互作用研究方面的应用进展。
2 计算机模拟方法简介 2.1 量子化学计算方法QC方法有从头计算法(ab initio)、密度泛函理论(DFT)方法及半经验法等类型[20]。ab initio是求解多电子体系问题的量子理论全电子方法, 不借助经验参数, 在理论和计算上都比较严格, 计算结果较为可靠[21]; Hartree-Fock(HF)法是最基本的从头计算方法, 采用单电子近似[22-23], 通常在其基础上继续考虑电子相关校正; Moller-Plesset多体微扰理论(MP)是基于HF单行列式的电子相关方法, 该方法把单电子Fock算符之和组成零级哈密顿, 当计入微扰到
运用分子间相互作用能(
| $\begin{eqnarray} \text{Δ}E=E_{\text{s}}-∑\limits_{i}E_{\text{i}} \end{eqnarray}$ | (1) |
式中,
分子力场是由一套原子尺度的势函数和力常数构成的势能场, 它是分子力学甚至整个分子模拟方法的核心。一个分子力场由分子内相互作用和分子间相互作用两大部分构成, 总能量(
| $\begin{eqnarray} E_{\text{total}}=E_{\text{bond}}+E_{\text{angle}}+E_{\text{torsion}}+E_{\text{vdW}}+E_{\text{Hbond}}+E_{\text{e}}+E_{\text{dipole}} \end{eqnarray}$ | (2) |
分子力场的参数由第一性原理计算结果或者实验结果拟合得到。目前分子力场已经从MM、AMBER、CHARM等第一代力场, CFF系列、PCFF等第二代力场, 发展到COMPASS从头算力场、ReaxFF反应力场等先进力场[27]。MM方法基于比较简单的模型建立分子力场公式进行计算, 计算公式中采用了大量的经验参数, 因此大大简化了计算过程, 与ab initio和半经验方法相比, 用MM计算较大体系可以节省计算时间。
2.3 分子动力学模拟MD模拟方法的建立来自于MM的发展, 它是利用牛顿力学基本原理, 通过求解运动方程得到所有原子的运动轨迹, 进而基于轨迹计算得到所需各种性质的一种方法, 适用于研究复杂凝聚态物质。经典MD模拟是在特定分子力场下, 通过运用力、速度和位置等参数动态模拟材料的结构和性能; 从头算分子动力学方法(AIMD)基于第一性原理, 将DFT方法与MD方法有机结合起来, 是计算机模拟中最重要的方法之一[28]。
2.4 耗散粒子动力学模拟DPD是在MD等方法的基础上发展而来的, 已经被广泛应用在多种复杂体系的模拟当中。DPD模拟方法基于粗粒化模型, 形象地说, 就是把一条高分子链模拟为一串“珠子”, 用每一个“珠子”代表一小块区域的集体行为, 并且在粒子间引入了“软”的相互作用势, 因此它可以用来模拟在较大尺度上系统的结构及演变。在DPD模拟中, 粒子的运动依然是遵循牛顿运动方程, 耗散力和随机力分别代表粒子之间的动力相互作用, 采用数值积分求解运动方程得到每个粒子在不同时刻的位置[27]。
3 计算机模拟在混合炸药分子间相互作用研究中的应用 3.1 对炸药聚合体分子间相互作用的研究在混合炸药体系中, 单质炸药的聚合体的分子间相互作用是较简单的情形。Politzer等[29]用DFT-B3PW91方法研究了二甲基硝胺的二聚体, 计算得到了其表面静电势图, 发现了两种二聚体结合方式, 并可由二聚体静电势推知第三个分子的结合方式, 对此聚合体的研究同样与二甲基硝胺晶体的结构相关(图 1)。李金山等运用ab initio方法在HF/6-31

通过量子化学计算可详细分析单质炸药聚合体中弱相互作用。宋华杰等[32]运用DFT结合SAPT方法研究了库仑力、色散力和交换排斥能等不同分子间力在TATB二聚体中所起的作用, 揭示了TATB分子间作用的本质。Chermahini等[33]采用DFT方法在B3LYP/6-31++G(d, p)水平下研究了四唑二聚体的分子间相互作用和氢键。陈天娜等采用DFT方法在B3LYP/6-31++
对炸药分子晶体的研究方面, Politzer等[38]采用QC方法在HF/6-311+

|
图 2 TATB晶体中分子间相互作用和 |
实际应用中, 常将不同种炸药混合使用以达到改进配方的目的, 因此不同炸药分子之间的相互作用也是混合体系中的常见情形。较为粗略但计算耗时较少的半经验方法有所应用, 李金山等[40]用半经验分子轨道(MO)方法在PM3水平上研究了TNB与TATB的分子间相互作用, 得到经色散能校正后的相互作用能。
DFT方法更为精确, 在研究不同炸药分子之间作用方面应用较多, 其计算方法与同种炸药分子间作用的计算类似。侯素青等[41]用DFT方法在B3LYP/6-31G(d)水平下研究了四种氮杂杯[4]芳烃类与RDX形成的复合体的结构, 分子间相互作用能由BSSE和ZPE校正进行计算, 并对四种复合体的相互作用能进行比较, 用NBO分析揭示了相互作用的本质, 结果表明复合体的相互作用能主要由氢键所贡献; 张文艳等[42]采取相同方法进一步对四种氮杂杯[6]芳烃主体单体及其与HMX形成的四种复合体进行了研究, 发现带有取代基的复合物的相互作用能大于没有带取代基的复合物, 带有氨基取代基的复合物的相互作用能大于带有硝基取代基的复合物。牛晓庆等[43]用DFT方法在B3LYP/6-31G(d)水平上研究了B炸药的主要成分TNT与RDX分子间的相互作用, 对于TNT+RDX混合体系校正后的相互作用能为:
| $\begin{eqnarray} \text{Δ}E_{\text{C}} =E_{\text{TNT}+\text{RDX}}-E_{\text{TNT}}-E_{\text{RDX}}+BSSE \end{eqnarray}$ | (3) |
式中,
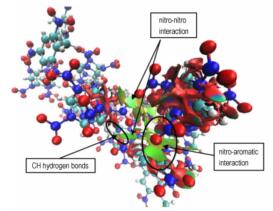
本课题组采用DFT方法在B3LYP/6-311++G(d, p)水平下计算了甲基硝基胍-硝酸肼低共熔物的分子间作用[44], 分析了结合能、氢键作用的贡献和分子聚集数的影响; 采用DFT-D计算方法得到了TNT/CL-20二聚体的最优化构型, 分析了其分子间相互作用(图 3), 并用MD模拟研究了TNT/CL-20共晶炸药的力学性能、稳定性和相互作用形式[45]。

|
图 3 TNT/CL-20共晶炸药中的分子间相互作用[45] Fig.3 Intermolecular interaction in TNT/CL-20 co-crystal explosive[45] |
对于含能组分与其他添加组分间的相互作用, 主要应用DFT方法进行研究。黄辉等[46]用DFT方法在B3LYP/6-31
此外, 针对添加改性组分常有高聚物而使体系较大计算量增大的困难, 半经验方法也得到应用。居学海[49]、孙小巧[50]、范晓薇等[51]用MO方法在PM3水平上分别研究了三乙二醇二硝酸酯(TEGDN)、丁三醇三硝酸酯(BTTN)和硝化甘油(NG)与高分子粘结剂形成的含能共混体系, 在此水平下, 相互作用能(
| $\begin{eqnarray} \text{Δ}E=\text{Δ}E^{\text{PM}3}+\text{Δ}E^{\text{D}} \end{eqnarray}$ | (4) |
式中,色散能可以由原子-原子势经验方法计算即:
| $\begin{eqnarray} \text{Δ}E^{\text{D}}=-∑\limits^A_i∑\limits^B_jC_{\text{ij}}R^{-6}_{\text{ij}} \end{eqnarray}$ | (5) |
式中,
分子模拟技术也被应用于炸药和其他组分的分子间相互作用研究, Cumming等[52]采用MM和MD模拟方法研究了(HMX+PNMO)超分子体系的结构和HMX与PNMO间的相互作用。孙小巧等[53]运用MM方法研究典型高能炸药(HMX, RDX, CL-20)与高分子粘结剂(PEG, HTPB, GAP, AMMO, BAMO等)混合物的分子间相互作用, 计算了上述超分子体系的结构与结合能, 发现相互作用能随高分子聚合度的增加而变化。以上计算结果可以为相关含能组分与高分子粘结剂的配方设计提供理论参考。
3.4 对添加改性组分分子间相互作用的研究探索粘结剂、增塑剂、固化剂等添加剂组分之间, 特别是组分与聚合物间的相互作用, 对于深入研究改性机理及筛选合理的混合炸药配方具有重要意义。张艳丽等[54]运用DFT法在B3LYP/6-31G水平上研究了高聚物粘结剂与硅烷偶联剂的混合体系, 发现高聚物粘结剂与硅烷偶联剂之间存在较强的电荷转移, 分子间存在H…O和F…H等弱氢键作用, 成功地模拟了高分子链与增塑剂的相互作用情形。
混合炸药中添加改性组分体系大多数是复杂凝聚态混合物, 组分相容性问题及介观结构性能等也为我们所关注, 更深入的分子模拟和热力学分析是必要的。特别是对于含聚合物的添加组分体系, 由于不容易用实验测定其升华热等热力学性质, 可以运用MD模拟, 选用包括径向分布函数、内聚能密度和溶度参数等来表征其相互作用情况。径向分布函数
| $\begin{eqnarray} E_{\text{coh}}=\text{Δ}H_{\text{V}}-RT \end{eqnarray}$ | (6) |
| $\begin{eqnarray} CED=E_{\text{coh}}/V \end{eqnarray}$ | (7) |
式中,
| $\begin{eqnarray} δ=\sqrt{E_{\text{coh}}/V} \end{eqnarray}$ | (8) |
当两种组分共混时, 其混合热的Hildebrand半经验式可表示为:
| $\begin{eqnarray} \frac{\text{Δ}H_{\text{m}}}{Vφ_{1}φ_{2}}=(δ_{1}-δ_{2})^{2} \end{eqnarray}$ | (9) |
式中,
按照热力学的自由能判据, 自发过程进行的必要条件是自由能变化小于零, 即:
| $\begin{eqnarray} \text{Δ}G_{\text{m}}=\text{Δ}H_{\text{m}}-T\text{Δ}S_{\text{m}}<0 \end{eqnarray}$ | (10) |
式中,

|
图 4 含能粘结剂PGN及其与增塑剂共混物的优化分子模型[61] Fig.4 Optimized models of energetic binder PGN and its blend with plasticizer[61] |
组分界面间相互作用问题的难点在于涉及到介观结构。混合炸药的力学、热力学等性质通常是以宏观材料测试方法进行研究[11-13], 作为复杂混合体系, 其介观尺度测试有一定困难。然而介观尺度结构对有关性能具有关键影响, 寻找一个有效的微观模型与宏观性能的关联方法成为一个难题, 而DPD方法则提供了一个有效的解决手段。
张艳丽等用DPD方法研究了TATB基PBX中F2311、F2312、F2313和F2314的聚合行为, 发现四种不同的含氟聚合物都形成网-球状结构, 从而包裹住TATB炸药分子, 通过与不同量的氯三氟乙烯(CTFE)混合的模拟, 发现随着CTFE量增加含氟聚合物发生相分离, 并发现在350 K和400 K含氟聚合物分别形成了网状和蜂窝状结构[62]; 同样采用DPD模拟方法研究了在TATB基PBX中硅烷偶联剂的有关行为, 发现了一种新的偶联机理, 即与TATB亲和的粘合剂结构单元在界面上组装, 与TATB不亲和的粘合剂结构单元则通过皱缩在其中的硅烷偶联剂而粘合在一起, 从而提高粘合剂和TATB的粘合作用[63]。周阳等采用DPD方法系统地研究了含能聚合物自组装及其与纳米含能材料的相互作用问题:通过研究键角势弯曲系数对模拟体系温度控制的影响建立了自定义的温差参数
| $\begin{eqnarray} δ_{\text{T}}=aφ^{\text{b}}\text{Δ}^{\text{c}}_{\text{t}} \end{eqnarray}$ | (11) |
式中,

|
图 5 纳米棒对含能均聚物两相界面行为的影响[67] Fig.5 Effect of nanorods on the interface behavior of energetic homopolymer[67] |
炸药体系中分子间相互作用的深入探讨, 对于进行混合炸药配方设计和性能研究, 具有指导作用。从描述电子结构和分子结构的ab initio方法、DFT方法和半经验分子轨道方法等量子化学计算方法, 到应用分子力场(或势函数)的MM、MD和DPD等分子模拟方法, 都是研究混合炸药体系中分子间相互作用的有效工具。计算机模拟混合炸药分子间作用有以下发展方向:
(1) 不断改进QC等计算方法, 提高计算准确率, 减少耗时。从基本的ab initio和DFT方法, 到相关泛函的改进和更多理论方法的建立, 使QC计算的准确性不断提升; 半经验法的应用使较大体系的计算成为可能, 同样的体系采用MM方法将进一步节省计算时间。
(2) 深入改进和发展相关的分子力场。力场是分子模拟技术的重要基础, QC方法的发展和相关理论的进步, 也将进一步带动相关力场的开发; 特定分子间作用势的研究, 将为建立适定力场提供基础, 也为相关力场的参数化修正提供了理论参考, 提高混合炸药体系分子模拟的适用性和准确性。
(3) 继续推进多尺度计算机模拟方法的贯通和发展。随着混合炸药结构性能研究的深入, 从微观到介观再到宏观研究的多尺度贯通问题亟待得到解决; 更多分子模拟方法如MC模拟、DPD模拟等的发展和应用, 将使混合炸药体系中多尺度问题得到解决。
总之, 随着混合炸药体系分子间相互作用研究的不断进展, 理论和实际应用中的更多的问题将得到解决。
致谢:感谢中物院化工材料研究所周阳老师对本文的指导。
| [1] |
舒远杰, 霍冀川著.
炸药学概论[M]. 北京: 化学工业出版社, 2011.
SHU Yuan-jie, HUO Ji-chuan. Introduction to explosives[M]. Beijing: Chemical Industry Press, 2011 |
| [2] |
马卿, 舒远杰, 罗观, 等. TNT基熔铸炸药:增韧增弹的途径及作用[J].
含能材料, 2012, 20(5): 618-629. MA Qing, SHU Yuan-jie, LUO Guan, et al. Toughening and elasticizing route of TNT based melt cast explosives[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(5): 618-629. |
| [3] |
李吉祯, 王炜, 刘芳莉, 等. 稳定剂对ADN和NC初期相互作用的影响[J].
火炸药学报, 2011, 34(2): 61-64. LI Ji-zhen, WANG Yi, LIU Fang-li, et al. Influences of stabilizers on the nascent interaction between AND and NC[J]. Chinese Journal of Explosives & Propellants, 2011, 34(2): 61-64. |
| [4] |
宋秀铎, 赵凤起, 王江宁, 等. BAMO-AMMO的热行为及其与含能组分的相容性[J].
火炸药学报, 2008, 31(3): 75-78. SONG Xiu-duo, ZHAO Feng-qi, WANG Jiang-ning, et al. Thermal behaviors of BAMO-AMMO and its compatibility with some energetic materials[J]. Chinese Journal of Explosives & Propellants, 2008, 31(3): 75-78. |
| [5] |
张腊莹, 衡淑云, 刘子如, 等. NC/NG与ADN的相互作用[J].
含能材料, 2009, 17(1): 95-98. ZHANG La-ying, HENG Shu-yun, LIU Zi-ru, et al. Interaction of NG/NC with AND[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(1): 95-98. |
| [6] |
张腊莹, 衡淑云, 刘子如, 等. PBT与高能氧化剂的相互作用的热分析法研究[J].
含能材料, 2009, 17(6): 668-672. ZHANG La-ying, HENG Shu-yun, LIU Zi-ru, et al. Interactions of PBT with some high energy oxidizers by thermal analysis[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(6): 668-672. |
| [7] |
王晓红, 衡淑云, 张皋, 等. DSC/TG-MS联用技术研究CL-20与NC-NG体系的相互作用[J].
火炸药学报, 2007, 30(4): 20-24. WANG Xiao-hong, HENG Shu-yun, ZHANG Gao, et al. Research on the interaction between CL-20 and NC-NG system via DSC/TG-MS[J]. Chinese Journal of Explosives & Propellants, 2007, 30(4): 20-24. |
| [8] |
何少蓉, 张林军, 衡淑云, 等. 量气法研究ADN与(NC+NG)的相互作用[J].
含能材料, 2008, 16(2): 225-228. HE Shao-rong, ZHANG Lin-jun, HENG Shu-yun, et al. Study on interaction of AND and (NC+NG) by gasometric method[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2008, 16(2): 225-228. |
| [9] |
何少蓉, 衡淑云, 张林军, 等. 量气法研究三种黏合剂与CL-20混合体系的热行为[J].
含能材料, 2010, 18(1): 37-41. HE Shao-rong, HENG Shu-yun, ZHANG Lin-jun, et al. Thermal behaviors of CL-20 systems mixed with three binders by gasometric method[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2010, 18(1): 37-41. |
| [10] |
岳璞, 衡淑云, 韩芳, 等. 三种方法研究ADN与几种粘合剂的相容性[J].
含能材料, 2008, 16(1): 66-69. YUE Pu, HENG Shu-yun, HAN Fang, et al. Compatibilities of AND with five kinds of binders[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2008, 16(1): 66-69. |
| [11] |
李江存, 焦清介, 任慧, 等. 不同键合剂与RDX表界面作用[J].
含能材料, 2009, 17(3): 274-277. LI Jiang-cun, JIAO Qing-jie, REN Hui, et al. Interfacial bonding between RDX and bonding agents[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(3): 274-277. |
| [12] |
周文静, 马亚南, 王克勇, 等. NTO与黏结剂的界面作用[J].
火炸药学报, 2010, 33(4): 40-43. ZHOU Wen-jing, MA Ya-nan, WANG Ke-yong, et al. Interfacial interaction between NTO and bindings[J]. Chinese Journal of Explosives & Propellants, 2010, 33(4): 40-43. |
| [13] |
倪冰, 覃光明, 冉秀伦. GAP/HTPB共混粘合剂体系的力学性能研究[J].
含能材料, 2010, 18(2): 167-173. NI Bing, QIN Guang-ming, RAN Xiu-lun. Mechanical properties of GAP/HTPB blend binders[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2010, 18(2): 167-173. |
| [14] |
潘碧峰, 罗运军, 谭惠民. CL-20与树形分子键合剂的粘附性能研究[J].
含能材料, 2004, 12(4): 199-202. PAN Bi-feng, LUO Yun-jun, TAN Hui-min. Study on interaction between CL-20 and dendritic bonding agent[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12(4): 199-202. |
| [15] |
潘碧峰, 罗运军, 谭惠民. 树形分子键合剂包覆AP及其相互作用研究[J].
含能材料, 2004, 12(1): 6-9. PAN Bi-feng, LUO Yun-jun, TAN Hui-min. Study on interaction between AP and dendritic bonding agent[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12(1): 6-9. |
| [16] |
潘碧峰, 罗运军, 谭惠民. 树形分子键合剂与HMX的相互作用[J].
火炸药学报, 2004, 27(3): 25-28. PAN Bi-feng, LUO Yun-jun, TAN Hui-min. Interaction between HMX and dendritic bonding agent[J]. Chinese Journal of Explosives & Propellants, 2004, 27(3): 25-28. |
| [17] |
张斌, 罗运军, 谭惠民. 多种键合剂与CL-20界面的相互作用机理[J].
火炸药学报, 2005, 28(3): 23-26. ZHANG Bin, LUO Yun-jun, TAN Hui-min. Interactional mechanism of interface between CL-20 and some bonding agents[J]. Chinese Journal of Explosives & Propellants, 2005, 28(3): 23-26. |
| [18] |
李玉斌, 李金山, 黄辉, 等. 新型硼酸酯键合剂与HMX的键合作用[J].
火炸药学报, 2010, 33(4): 36-39. LI Yu-bin, LI Jin-shan, HUANG Hui, et al. Interaction between a novel borate ester bonding agent and HMX[J]. Chinese Journal of Explosives & Propellants, 2010, 33(4): 36-39. |
| [19] |
Rohac M, Zeman S, Ruzicka A. Crystallography of 2, 2′, 4, 4′, 6, 6′-hexanitro-1, 1′-biphenyl and its relation to initiation reactivity[J].
Chemistry of Materials, 2008, 20(9): 3105-3109. DOI:10.1021/cm702829k |
| [20] |
Foresman J B, Frisch A.
Exploring chemistry with electronic structure methods(second edition)[M]. Gaussian Inc Pittsburgh PA, 1996 |
| [21] |
徐光宪, 黎乐民.
量子化学(基本原理和从头计算法)[M]. 北京: 科学出版社, 1984.
XU Guang-xian, LI Le-min. Quantum chemistry(basic principles and ab initio calculation method)[M]. Beijing: Science Press, 1984 |
| [22] |
Hartree D R. Wave mechanics of an atom with a non-coulomb central field[J].
Proc Camb Phil Soc, 1928, 24: 89-110. DOI:10.1017/S0305004100011919 |
| [23] |
Fock V Z. The initial degrees of freedoms of the electron[J].
Phys, 1931, 68: 522-534. DOI:10.1007/BF01391145 |
| [24] |
Moller C, Plesset M S. Note on an approximation treatment for many-electron systems[J].
Phys Rev, 1934, 46: 618-622. DOI:10.1103/PhysRev.46.618 |
| [25] |
Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].
Phys Rev B, 1988, 37: 785-789. DOI:10.1103/PhysRevB.37.785 |
| [26] |
肖鹤鸣, 居学海.
高能体系中的分子间相互作用[M]. 北京: 科学出版社, 2004.
XIAO He-min, JU Xue-hai. Intermolecular interactions in the energetic systems[M]. Beijing: Science Press, 2004 |
| [27] |
杨小震.
软物质的计算机模拟与理论方法[M]. 北京: 化学工业出版社, 2010.
YANG Xiao-zhen. Computer simulation and theoretical methods for soft matters[M]. Beijing: Chemical Industry Press, 2010 |
| [28] |
ClementiE, CorongiuG, 帅志刚, 等. 从原子到大分子体系的计算机模拟-计算化学50年[J].
化学进展, 2011, 23(9): 1795-1830. Clementi E, Corongiu G, Shuai Zhi-gang, et al. With computers from atoms to macromolecular systems[J]. Progress in Chemistry, 2011, 23(9): 1795-1830. |
| [29] |
Politzer P, Concha M C, Murray J S. Density functional study of dimers of dimethylnitramine[J].
International Journal of Quantum Chemistry, 2000, 80(2): 184-192. DOI:10.1002/(ISSN)1097-461X |
| [30] |
LI Jin-shan, XIAO He-ming, DONG Hai-shan. Theoretical study on intermolecular interaction of epoxyethane dimer[J].
Int J Quant Chem, 2000, 78: 94-98. DOI:10.1002/(ISSN)1097-461X |
| [31] |
LI Jin-shan, ZHAO Feng, JING Fu-qian. An ab initio study of intermolecular interactions of nitromethane dimer and nitromethane trimer[J].
Journal of Computational Chemistry, 2003, 24(3): 345-352. DOI:10.1002/(ISSN)1096-987X |
| [32] |
宋华杰, 肖鹤鸣, 董海山. TATB二聚体分子间作用力及其气相几何构型研究[J].
化学学报, 2007, 65(12): 1101-1109. SONG Hua-jie, XIAO He-ming, DONG Hai-shan. Intermolecular forces and gas geometries of TATB dimers[J]. Acta Chimica Sinica, 2007, 65(12): 1101-1109. DOI:10.3321/j.issn:0567-7351.2007.12.002 |
| [33] |
Chermahinia A N, Ghaedia A, Teimourib A, et al. Density functional theory study of intermolecular interactions of cyclic tetrazole dimers[J].
Journal of Molecular Structure: THEOCHEM, 2008, 867: 78-84. DOI:10.1016/j.theochem.2008.07.023 |
| [34] |
陈天娜, 汤业朋, 宋华杰. 氧化呋咱二聚体分子间相互作用的理论计算[J].
含能材料, 2007, 15(6): 641-645. CHEN Tian-na, TANG Ye-peng, SONG Hua-jie. Theoretical study on intermolecular interaction of furoxan dimers[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2007, 15(6): 641-645. |
| [35] |
陈天娜, 汤业朋, 肖鹤鸣. α-双环-HMX晶体中二聚作用的理论研究[J].
化学学报, 2010, 68(19): 1986-1990. CHEN Tian-na, TANG Ye-peng, XIAO He-min. A theoretical study on the dimerization of α-bicyclo-HMX[J]. Acta Chimica Sinica, 2010, 68(19): 1986-1990. |
| [36] |
胡银, 马海霞, 张教强, 等. 3, 6-二氨基-1, 2, 4, 5-四嗪二聚体分子间相互作用的理论研究[J].
化学通报, 2010(3): 263-268. HU Yin, MA Hai-xia, ZHANG Jiao-qiang, et al. Theoretical study on intermolecular interactions of 3, 6-diamino-1, 2, 4, 5-tetrazine dimers[J]. Chemistry Bulletin, 2010(3): 263-268. |
| [37] |
胡银, 邵颖慧, 胡荣祖, 等. BTATz二聚体分子间相互作用的理论计算[J].
含能材料, 2012, 20(3): 273-279. HU Yin, SHAO Ying-hui, HU Rong-zu, et al. Theoretical study on intermolecular interactions of BTATz dimers[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(3): 273-279. |
| [38] |
Politzer P, MA Yu-guang. Noncovalent intermolecular energetics: RDX crystal[J].
International Journal of Quantum Chemistry, 2004, 100(5): 733-739. DOI:10.1002/(ISSN)1097-461X |
| [39] |
ZHANG Chao-yang, WANG Xiao-chuan, HUANG Hui. π-Stacked interactions in explosive crystals: buffers against external mechanical stimuli[J].
J Am Chem Soc, 2008, 130(26): 8359-8365. DOI:10.1021/ja800712e |
| [40] |
LI Jin-shan, XIAO He-ming, DONG Hai-shan. A study on the intermolecular interaction of energetic system-mixtures containing -CNO2 and -NH2 groups[J].
Propellants, Explosives, Pyrotechnics, 2000, 25(1): 26-30. DOI:10.1002/(ISSN)1521-4087 |
| [41] |
侯素青, 曹端林, 张文艳, 等. 氮杂杯[J].
火炸药学报, 2008, 31(5): 19-23. HOU Su-qing, CAO Duan-lin, ZHANG Wen-yan, et al. Density functional theory of intermolecular interactions of aza-calix[4]arene with RDX[J]. Chinese Journal of Explosives & Propellants, 2008, 31(5): 19-23. |
| [42] |
张文艳, 曹端林, 侯素青, 等. 氮杂类杯[6]芳烃与HMX分子间相互作用的理论研究[J].
含能材料, 2009, 17(4): 436-441. ZHANG Wen-yan, CAO Duan-lin, HOU Su-qing, et al. Theoretical studies on intermolecular interactions between azacalix[6]arene and HMX[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2009, 17(4): 436-441. |
| [43] |
牛晓庆, 张建国, 冯晓军, 等. B炸药主要组分TNT和RDX分子间相互作用的理论研究[J].
化学学报, 2011, 69(14): 1627-1638. NIU Xiao-Qing, ZHANG Jian-Guo, FENG Xiao-Jun, et al. Theoretical investigation on intermolecular interactions between the ingredients TNT and RDX of composition B[J]. Acta Chim Sinica, 2011, 69(14): 1627-1638. |
| [44] |
陈玲, 李华荣, 熊鹰, 等. 甲基硝基胍-硝酸肼低共熔物结构及分子间作用[J].
含能材料, 2012, 20(5): 560-564. CHEN Ling, LI Hua-rong, XIONG Ying, et al. Structure and molecular interaction of methyl-nitroguanidine and hydrazine nitrate eutectics[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(5): 560-564. |
| [45] |
LI Hua-rong, SHU Yuan-jie. Theoretical insights into the nature of intermolecular interactions in TNT/CL-20 cocrystal and its properties[C]∥New Trends in Research of Energetic Materials, Czech Republic, 2013, 742.
|
| [46] |
黄辉, 李金山. HMX与含硼化合物相互作用的理论计算[J].
原子与分子物理学报, 2007, 24(1): 106-110. HUANG Hui, LI Jin-shan. Theoretical calculation of interaction between HMX and B-containing compound[J]. Journal of Atomic and Molecular Physics, 2007, 24(1): 106-110. |
| [47] |
林小雄, 王明良, 赵凤起, 等. 硝基甲烷与氨基及羟基化合物间的相互作用[J].
火炸药学报, 2012, 35(4): 1-4. LIN Xiao-xiong, WANG Ming-liang, ZHAO Feng-qi, et al. Interaction between nitromethane and amino, hydroxyl compounds[J]. Chinese Journal of Explosives & Propellants, 2012, 35(4): 1-4. |
| [48] |
Venkatesan V, Polke B G, Sikder A K. Ab initio study on the intermolecular interactions between 1, 1-diamino-2, 2-dinitroethylene and acetylene: pull effect on complex formation[J].
Computational and Theoretical Chemistry, 2012, 995: 49-54. DOI:10.1016/j.comptc.2012.06.028 |
| [49] |
居学海, 范晓微, 孙小巧, 等. 三乙二醇二硝酸酯与高分子黏结剂在混合体系中的分子间相互作用[J].
化学推进剂与高分子材料, 2007, 5(5): 44-47. JU Xue-hai, FAN Xiao-wei, SUN Xiao-qiao, et al. Intermolecular interaction between triethylene glycol dinitrate and polymer binders in the mixed systems[J]. Chemical Propellants & Polymeric Materials, 2007, 5(5): 44-47. |
| [50] |
孙小巧, 范晓薇, 居学海, 等. 丁三醇三硝酸酯与高分子黏合剂的相互作用[J].
火炸药学报, 2007, 30(3): 1-4. SUN Xiao-qiao, FAN Xiao-wei, JU Xue-hai, et al. Interactions between BTTN and polymer binders[J]. Chinese Journal of Explosives & Propellants, 2007, 30(3): 1-4. |
| [51] |
范晓薇, 居学海, 孙小巧, 等. 硝化甘油与高分子黏合剂混合体系相互作用的理论研究[J].
火炸药学报, 2009, 32(3): 46-49. FAN Xiao-wei, JU Xue-hai, SUN Xiao-qiao, et al. Theoretical studies of interaction of complex system nitroglycerin-polymer binder[J]. Chinese Journal of Explosives & Propellants, 2009, 32(3): 46-49. |
| [52] |
Cumming A S, Leiper G A, Robson E. Molecular modeling as a tool to aid the design of polymer bonded exploxives[C]∥24th International Annual Conference of ICT, Germany: Karlsruhe, 1993.
|
| [53] |
孙小巧. 高能氧化剂与粘合剂的分子间相互作用[D]. 南京: 南京理工大学, 2007.
SUN Xiao-qiao. Intermolecular interactions between energetic oxidizers and binders[D]. Nanjing: Nanjing University of Science and Technology, 2007. |
| [54] |
张艳丽, 姬广富, 龚自正. 高聚物粘结剂与硅烷偶联剂分子间相互作用[J].
南京理工大学学报(自然科学版), 2009, 33(5): 682-686. ZHANG Yan-li, JI Guang-fu, GONG Zi-zeng. Intermolecular interactions of polymeric binders and silane coupling agents[J]. Journal of Nanjing University of Science and Technology(Natural Science), 2009, 33(5): 682-686. |
| [55] |
符若文, 李谷, 冯开才.
高分子物理[M]. 北京: 化学工业出版社, 2005.
FU Ruo-wen, LI Gu, FENG Kai-cai. Physics of high polymers[M]. Beijing: Chemical Industry Press, 2005 |
| [56] |
李倩, 姚维尚, 谭惠民. 叠氮粘合剂与硝酸酯溶度参数的分子动力学模拟[J].
含能材料, 2007, 15(4): 370-373. LI Qian, YAO Wei-shang, TAN Hui-min. Molecular dynamics simulation of solubility parameter of azide binders and nitrate ester[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2007, 15(4): 370-373. |
| [57] |
Abou-Rachid H, Lussier L S, Ringuette S, et al. On the correlation between miscibility and solubility properties of energetic plasticizers/polymer blends: modeling and simulation studies[J].
Propellants, Explosives, Pyrotechnics, 2008, 33(4): 301-310. DOI:10.1002/prep.v33:4 |
| [58] |
杨月诚, 焦东明, 强洪夫, 等. HTPB推进剂组分溶度参数的分子模拟研究[J].
含能材料, 2008, 16(4): 191-195. YANG Yue-cheng, JIAO Dong-ming, QIANG Hong-fu, et al. Molecular simulation of solubility parameter for HTPB solid propellants[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2008, 16(4): 191-195. |
| [59] |
付一政, 刘亚青, 兰艳花. 端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟[J].
物理化学学报, 2009, 25(7): 1267-1272. FU Yi-zheng, LIU Ya-qing, LAN Yan-hua. Molecular dynamics simulation on compatibility of hydroxyl-terminated polybutadiene/plasticizer blends[J]. Acta Phys Chim Sin, 2009, 25(7): 1267-1272. |
| [60] |
黄锐, 姚维尚, 谭惠民. 叠氮纤维素结构和溶度参数的分子模拟[J].
含能材料, 2008, 16(4): 446-449. HUANG Rui, YAO Wei-shang, TAN Hui-min. Molecular simulation on structure and solubility parameter of azidodeoxycellulose[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2008, 16(4): 446-449. |
| [61] |
QIAN Wen. Simulation study on the miscibility of energetic binder poly(glycidyl nitrate) with several plasticizers[C]∥ Proceeding of the 6th International Conference of Molecular Simulations & Applied Informatics Technologies, Nanjing, 2012: 457-467.
|
| [62] |
ZHANG Yan-li, JI Guang-fu, ZHAO Feng, et al. Mesoscopic simulation of aggregate behaviour of fluoropolymers in the TATB-based PBX[J].
Molecular Simulation, 2011, 37(3): 237-242. DOI:10.1080/08927022.2010.543969 |
| [63] |
ZHANG Yan-li, JI Guang-fu, GONG Zi-zheng. New coupling mechanism of the silane coupling agents in the TATB-based PBX[J].
Molecular Simulation, 2013, 39(5): 423-427. DOI:10.1080/08927022.2012.738295 |
| [64] |
ZHOU Yang, LONG Xin-ping, ZENG Qing-xuan. Effect of the angular potential on the temperature control in dissipative particle dynamics simulations[J].
Molecular Simulation, 2012, 38(12): 961-969. DOI:10.1080/08927022.2012.679618 |
| [65] |
ZHOU Yang, LONG Xin-ping, ZENG Qing-xuan. Simulation studies of the interfaces of incompatible glycidyl azide polymerhydroxyl-terminated polybutadiene blends by dissipative particle dynamics[J].
Journal of Applied Polymer Science, 2012, 125(2): 1530-1537. DOI:10.1002/app.v125.2 |
| [66] |
ZHOU Yang, LONG Xin-ping, ZENG Qing-xuan. Simulation study of the morphologies of energetic block copolymers based on glycidyl azide polymer[J].
Journal of Applied Polymer Science, 2013, 129(1): 480-486. DOI:10.1002/app.v129.1 |
| [67] |
ZHOU Yang, LONG Xin-ping, ZENG Qing-xuan. Dissipative particle dynamics studies on the interface of incompatible A/B homopolymer blends in the presence of nanorods[J].
Polymer, 2011, 52(26): 6110-6116. DOI:10.1016/j.polymer.2011.10.052 |
| [68] |
ZHOU Yang, LONG Xin-ping, ZENG Qing-xuan, et al. A novel nanocage from the cooperative self-assembly of coil-rod-coil triblock copolymers and nanopartilces[J].
Macromolecular Rapid Communications, 2013, 34(10): 883-886. DOI:10.1002/marc.201300001 |
Computer simulation methods such as quantum chemistry calculation, molecular mechanics, molecular dynamics and dissipative particle dynamics were introduced.