




2. 氟氮化工资源高效开发与利用国家重点实验室, 陕西 西安 710065
2. State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi′an 710065, China
追求高能量、高密度是含能化合物合成研究领域永恒的主题, 欧美等西方国家以致密的张力环和笼型结构为母体结构单元, 引入—NO2等含能基团设计、合成出了八硝基立方烷(ONC)[1]和六硝基六氮杂异伍兹烷(CL-20)[2]等高能量密度化合物。上述含能化合物存在合成步骤多、合成难度大、制造成本和感度较高等突出问题, 严重制约其在武器装备中的推广应用。近年来, 基于呋咱环具有生成焓较高、热稳定性好的特点, 开展呋咱含能化合物设计、合成及性能研究已成为含能材料领域的研究热点之一[3-7]。Luk′yanov等[8-12]利用氨基呋咱中氨基和亚硝基化合物的氧化偶联反应构建出α-烷基取代的氧化偶氮基团, 再利用硝化、氟化反应进一步在烷基上引入含能基团, 已成功合成出多种结构新颖的α-多硝甲基氧化偶氮呋咱含能化合物, 其中, 以二氨基氧化偶氮呋咱为原料, 经过多步反应合成出3, 3′-双(三硝甲基氧化偶氮基)-4, 4′-氧化偶氮呋咱, 文献[11]只报道了该化合物的合成方法和熔点(64~66 ℃)。目前, 多硝甲基氧化偶氮呋咱化合物的设计、合成研究受到国内外含能材料领域的关注[13], 但已有化合物的爆轰性能和安全性能等应用基础性能尚未披露, 也未见文献对比研究多硝甲基氧化偶氮基团对呋咱化合物爆轰和安全性的影响规律。
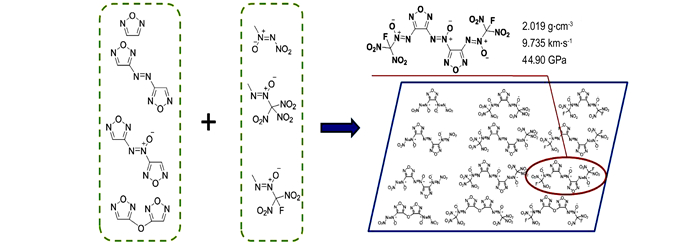
为了考察三硝甲基氧化偶氮基团和氟二硝甲基氧化偶氮基团对呋咱化合物性能的影响, 并与硝基氧化偶氮基团进行对比。本研究利用量子化学中的密度泛函理论, 对由呋咱(F)、偶氮呋咱(AF)、氧化偶氮呋咱(AOF)和呋咱醚(FE)四类呋咱母体结构和硝基氧化偶氮基(Ⅰ)、三硝甲基氧化偶氮基(Ⅱ)、氟二硝甲基氧化偶氮基(Ⅲ)三种含能基团组合而成的十二种呋咱化合物(图 1)的性能进行理论研究, 考察母体结构和含能基团对化合物密度、生成焓、爆速、爆压、撞击感度的影响, 为高能量密度化合物的设计、合成提供参考。

|
图 1 12种呋咱含能化合物结构 Fig.1 Structures of 12 kinds of furazan energetic compounds |
采用密度泛函理论(DFT)的B3LYP方法[14], 在6-31G**基组水平上对化合物结构进行了全优化, 经振动频率分析发现无虚频, 表明优化结构为势能面上的极小点。采用Multwfn程序[15]对各化合物的静电势进行统计分析, 获得静电势参数。采用Monte-Carlo法[16]计算了100次三种分子的体积, 取其平均值为摩尔体积(Vm), 并利用B.M.Rice等人[17]提出的公式和参数进行校正(式1), 获得化合物的密度(ρ)。
为了获得较准确的气相生成焓值, 设计了目标化合物的等键反应, 如图 2。利用反应焓变和其它小分子的气相生成焓值, 计算目标化合物的气相生成焓; 其中等键反应方法涉及到的其它小分子化合物的气相生成焓值, 采用G3方法[18]按照原子化方案计算得出。化合物的升华焓利用Politzer等人[19]提出的公式(式2)和化合物的静电势参数计算得出, 进而求得固相生成焓(ΔfH(s, M, 298K)Θ)(式3)。

|
图 2 设计的12种呋咱含能化合物等键反应 Fig.2 Designed isodesmic reactions for 12 kinds of furazan energetic compounds |
将含能化合物的组成、密度和生成焓数据代入Kamlet-Jacobs公式[20](式4、式5), 可计算出理论爆速(D)和理论爆压(p)。
分子结构中最弱键的键离解能(BDE)按照式6进行计算, 以H50表示的撞击感度按照与静电势参数有关的经验公式[21](式7)进行计算。
| $ \rho = 1.00462 \times \left( {\frac{M}{{{V_M}}}} \right) + 0.0021 \times \left( {\nu {\sigma _{{\rm{tot}}}}^2} \right) - 0.1586 $ | (1) |
| $ {\Delta _{{\rm{sub}}}}H = 0.000475 \times {A^2} + 2.1194 \times (\nu \sigma _{_{{\rm{tot}}}}^2)0.5-2.25 $ | (2) |
| $ {\Delta _{\rm{f}}}H_{\left({{\rm{s,M,298K}}} \right)}^\Theta = {\Delta _{\rm{f}}}H_{\left({{\rm{g,M,298K}}} \right)}^{\rm{\Theta }}-{\Delta _{{\rm{sub}}}}H $ | (3) |
| $ p = 1.558{\rho ^2}N{{\bar M}^{1/2}}{Q^{1/2}} $ | (4) |
| $ D = 1.01(N{{\bar M}^{1/2}}{Q^{1/2}})1/2\left({1 + 1.30\rho } \right) $ | (5) |
| $ BDE\left({A-B} \right) = H\left(A \right) + H\left(B \right)-H(A-B) $ | (6) |
| $ {H_{50}} =-0.0064\sigma _ + ^2 + 241.42\nu-3.43 $ | (7) |
12种呋咱含能化合物的稳定构型如图 3所示。由图 3可见, 呋咱环上所有的键长介于单键和双键键长之间, 呋咱环中存在较为明显的共振结构, 与其较高的稳定性较为一致。所有偶氮相连的较大基团均为反式排布, 为较为稳定的构型。偶氮键和氧化偶氮键均和与其直接相连的原子构成平面, 表明偶氮键有助于提高分子内的电子共轭作用, 从而分散电荷, 提高含能化合物的稳定性。以氧化偶氮呋咱化合物为例, AOF-Ⅰ的所有偶氮基的N=N键键长约为1.28 nm, 偶氮基和呋咱环之间的C—N键键长介于1.37~1.44 nm, 而偶氮基和硝基之间的N—N键键长约为1.49 nm, 键长较长, 受外界条件刺激, 可能为较易断裂的键; AOF-Ⅱ分子中, 偶氮基和三硝甲基的碳原子之间的C—N键键长约为1.49 nm, 而碳原子和硝基之间的C—N键长约为1.55 nm, 表明, C—NO2较C—N(O)=N更易断裂; AOF-Ⅲ中氟原子对C—N键的键长影响较小, C—NO2和C—N(O)=N中C—N键键长分别约为1.50 nm和1.55 nm。AOF-Ⅰ中硝基氧化偶氮基团中由N—N(—O)=N构成的平面和呋咱环的夹角为47.8°, AOF-Ⅱ和AOF-Ⅲ中由C—N(—O)=N构成的平面和呋咱环的夹角接近0°, 可见, 多硝甲基氧化偶氮基团和呋咱环具有较好的电子共振作用, 多硝甲基氧化偶氮呋咱化合物在稳定性方面可能优于硝基氧化偶氮呋咱化合物。

|
图 3 几何构型 Fig.3 Geometric configuration |
分子静电势和分子反应性、分子间相互作用具有重要的相关性, 通过可视化的静电势图可对化合物的含能性质进行定性判断, 同时, 通过统计分析, 获得静电势参数可进一步用于计算化合物的密度、生成焓、感度等性能。
在B3LYP/6-31G**水平下, 得到了化合物在0.001 electron·bohr-3电子密度等值面上的三维静电势分布示意图(图 4以氧化偶氮呋咱化合物为例), 并利用Multiwfn3.0.1程序, 对正负静电势区域的面积和强度进行了统计, 结果列于表 1。从图 4可见, 化合物的正静电势主要分布在中心呋咱环和桥联的偶氮基团上, 负静电势主要分布在周围的硝基和氧化偶氮基团的氧原子。Klapötke等人[22]认为在含能体系中, 正静电势区域所占的面积比例要大, 而且强度要大于负静电势区域。由表 1可知, 12种呋咱含能化合物的正静电势区域的面积和强度均大于负静电势区域的面积和强度, 表现出含能材料所特有的静电势分布特性。

|
图 4 AOF-Ⅰ、AOF-Ⅱ和AOF-Ⅲ的静电势图 Fig.4 Electrostatic potential of AOF-Ⅰ, AOF-Ⅱ and AOF-Ⅲ |
| 表 1 12种呋咱含能化合物静电势参数 Tab.1 Electrostatic potential parameters for 12 kinds of urazan energetic compounds |
对12种呋咱化合物的理论密度(ρ)、固相生成焓(ΔfH(s, M, 298K)Θ)、氧平衡(OB)、理论爆速(D)、理论爆压(p)进行了理论计算, 结果见表 2和图 5。
| 表 2 12种呋咱含能化合物的能量性能 Tab.2 Energy performances of 12 kinds of furazan energetic compounds |

|
图 5 呋咱化合物的密度、生成焓、氧平衡和爆速 Fig.5 Densities, enthalpies of formation, oxygen balances and detonation velocities of furazan compounds |
由表 2和图 5中数据可见, 将12种呋咱含能化合物密度的计算值和AF-Ⅰ、FE-Ⅰ的实测值[23-24]进行对比, 发现误差<3%, 表明所采用的计算方法可靠。对于每种桥联结构的呋咱化合物而言, 通过将硝基和氧化偶氮进行组合构建的含能基团均具有较高的密度, 且存在如下顺序:Ⅲ>Ⅱ>Ⅰ, 和硝基氧化偶氮基团相比较, 三硝甲基氧化偶氮基团可使呋咱化合物的密度提高约0.1 g·cm-3, 以氟替代硝基, 可使密度略有提高。可见, 在呋咱环上构建三硝甲基氧化偶氮基团和氟二硝甲基氧化偶氮基团, 是一种设计合成密度大于2.0 g·cm-3高能量密度化合物的有效途径。通过对比不同的桥联结构, 发现, 通过氧化偶氮基团相连的呋咱化合物普遍具有较高的密度, 该结果和Shreeve等人[23]的研究结果较为一致。
呋咱环自身的生成焓较高, 以其为结构单元构建的含能化合物均有较高的生成焓, 由图中结果可见, 12种化合物的生成焓均大于150 kJ·mol-1, 储存的化学能较高, 有助于提高爆轰过程中的爆热。通过对比, 可以发现桥联结构与生成焓的大小关系顺序为AF>AOF>FE>F; 含能基团与生成焓的大小关系顺序为Ⅰ>Ⅱ>Ⅲ。
三种含能基团均具有较高的氧含量, 使得12种呋咱化合物的氧平衡均≥0, 桥联结构与氧平衡的大小关系顺序为F>FE≈AOF>AF; 含能基团与氧平衡的大小关系顺序为Ⅱ>Ⅲ≈Ⅰ。其中, F-Ⅱ的氧平衡高达40.73%, 高于目前常用的高氯酸铵(AP, 27.23%)、二硝酰胺铵(ADN, 25.80%)、硝酸铵(AN, 20.00%)、硝仿肼(HNF, 13.00%)等离子型氧化剂, 是一种含氧量极高的分子型氧化剂。
爆速和爆压是含能化合物爆轰性能重要的表观参数, 受密度、生成焓、氧平衡以及分子组成的影响。含能基团与爆速、爆压的大小关系顺序为Ⅲ>Ⅰ>Ⅱ, 氟二硝甲基氧化偶氮基团构建的含能化合物均表现出较高的爆轰性能, AOF-Ⅲ的爆速高达9.735 km·s-1, 爆压高达44.90 GPa, 能量水平高于CL-20。含能基团中具有最高氧平衡的Ⅱ反而使爆速急剧下降, 同样, 氧平衡普遍较高的呋咱(F)结构化合物的爆速也较低, 可能是其较高的氧含量使得爆轰过程中产生二氧化碳、水等完全氧化产物, 虽然放热较大, 但是做工介质较少, 难以产生较高的爆轰作用, 而且, 大量氧元素剩余, 难以发挥其能量潜能, 将其和氧含量较低的含能材料配合使用, 有可能进一步提高爆轰性能。
3.4 安全性能含能化合物是否具有可接受的安全性能将是影响其应用的关键因素, 考察了含能基团和桥联结构对呋咱含能化合物的热稳定性和撞击感度的影响。
判断含能化合物热稳定性较为常用的一种方法是比较具有最小键级的化学键的键离解能(BDE)。对12种呋咱化合物优化构型的自然键级轨道(NBO)分析得到具有最小键级的化学键, 并分别计算了这些化学键的BDE, 计算结果列于表 3和图 6a中。
| 表 3 呋咱化合物最弱键及键离解能 Tab.3 The weakest bonds and corresponding BDEs of furazan compounds |

|
图 6 呋咱化合物的键离解能和撞击感度 Fig.6 BDEs and impact sensitivities of furazan compounds |
由表 3和图 6a可见, 12种呋咱化合物中的最弱键均为和硝基直接相连的化学键, 而且桥联结构对BDE的影响较小, 含能基团与BDE大小顺序关系为Ⅲ>Ⅱ>Ⅰ。硝基氧化偶氮基团具有最低的BDE, BDE约为117~119 kJ·mol-1, 三硝甲基氧化偶氮基团和氟二硝甲基氧化偶氮基团中, 通过引入碳原子, 构建了具有较稳定化学键的C—N键, 热稳定性得以改善, 同时, 以一个氟原子取代硝基后使稳定性进一步提高。根据肖鹤鸣等[24]提出的判断高能量密度化合物稳定性的定量标准, 若BDE>120 kJ·mol-1, 则认为满足品优高能量密度化合物的稳定性要求。三硝甲基氧化偶氮呋咱及氟二硝甲基氧化偶氮呋咱化合物的BDE均大于120 kJ·mol-1, 而且, 氟二硝甲基氧化偶氮呋咱化合物具有高于三硝甲基氧化偶氮呋咱的热稳定性。
根据静电势参数计算出12种呋咱化合物的撞击感度[21], 以特性落高H50表示, H50值越大, 撞击感度越低, 结果见表 3和图 6b。结果表明, 以氧化偶氮进行桥联的呋咱化合物具有较高的特性落高值, 安全性能较好。硝基氧化偶氮基团会提高呋咱化合物的撞击感度, 而氟二硝甲基氧化偶氮基团则有助于降低呋咱化合物的撞击感度。通过和BDE数据进行比较, 可见, 具有相同呋咱母体结构的化合物, 最弱键的断裂难易程度和撞击感度具有一定的相关性, BDE值越大, 撞击感度越低。AOF-Ⅲ的BDE值为157.55 kJ·mol-1, H50为36 cm, 表现出较好的热稳定性和低于RDX(H50为26 cm)[21]的撞击感度性能。
4 结论(1) 呋咱环和硝基、多硝甲基分别处于偶氮键的反式位置, 为较为稳定的构型; 和硝基直接相连的N—N键和C—N键的键长明显大于其它化学键, 可能为较易断裂的键; 多硝甲基氧化偶氮基团和呋咱环的共面性优于硝基氧化偶氮基团, 表明其稳定性较佳。
(2) 正静电势主要分布在中心呋咱环和桥联的偶氮基团上, 负静电势主要分布在周围的硝基和氧化偶氮基团的氧原子, 且正静电势区域的面积和强度均大于负静电势区域的面积和强度, 表现出含能材料所特有的静电势分布特性。
(3) 对于四种呋咱母体结构, 三硝甲基氧化偶氮基团及氟二硝甲基氧化偶氮基团均可大幅提高化合物的密度和氧平衡, 氟二硝甲基氧化偶氮呋咱化合物具有较高的爆速(>9400 m·s-1)和爆压(>41 GPa), 能量水平较高, 热稳定性和撞击感度也优于硝基氧化偶氮呋咱和三硝甲基氧化偶氮呋咱化合物。
(4) 理论计算结果表明, 3, 3′-双(氟二硝甲基氧化偶氮基)-4, 4′-氧化偶氮呋咱(AOF-Ⅲ)的密度为2.019 g·cm-3, 氧平衡为6.72%, 爆速为9.735 km·s-1, 爆压为44.90 GPa, 最弱键键离解能为157.55 kJ·mol-1, 特性落高为36 cm, 表现出优良的综合性能, 是一种具有应用潜力的高能量密度材料。
| [1] |
邱玲, 许晓娟, 肖鹤鸣. 多硝基立方烷的合成、结构和性能研究进展[J].
含能材料, 2005, 13(4): 262-268. QIU Ling, XU Xiao-juan, XIAO He-ming. Review on synthesis, structure and performance of polynitrocubanes[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2005, 13(4): 262-268. |
| [2] |
庞思平, 申帆帆, 吕芃浩, 等. 六硝基六氮杂异伍兹烷合成工艺研究进展[J].
兵工学报, 2014, 5: 725-732. PANG Si-ping, SHENG Fan-fan, LV Fan-hao, et al. Research progress in synthesis of hexanitrohexaazaisowurtzitane[J]. Acta Armamentarii, 2014, 5: 725-732. |
| [3] |
何玲, 董琳琳, 张光全, 等. 呋咱并[3, 4-e]-1, 2, 3, 4-四嗪-1, 3-二氧化物(FTDO)分子结构及性质的理论研究[J].
含能材料, 2012, 20(6): 693-696. HE Ling, DONG Lin-lin, ZHANG Guang-quan, et al. Structure and properties of furazano[3, 4-e]-1, 2, 3, 4-tetrazine-1, 3-dioxide[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2012, 20(6): 693-696. |
| [4] |
翟连杰, 王伯周, 霍欢, 等. 3, 4-双(3-硝基呋咱-4-氧基)呋咱合成、晶体结构及热性能[J].
含能材料, 2015, 23(1): 18-22. ZHAI Lian-jie, WANG Bo-zhou, HUO Huan, et al. Synthesis, crystal structure and thermal behavior of 3, 4-Bis (3-nitrofurazan-4-oxy) furazan[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2015, 23(1): 18-22. DOI:10.11943/j.issn.1006-9941.2015.01.004 |
| [5] |
刘宁, 王伯周, 李辉, 等. 两种呋咱并[3, 4-b]四唑并[1, 2-d]吡嗪化合物的合成、晶体结构及热性能[J].
含能材料, 2015, 23(1): 13-17. LIU Ning, WANG Bo-zhou, LI Hui, et al. Synthesis, crystal structure and thermal properties of two furazano[3, 4-b] tetrazolo[1, 2-d] pyrazines[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2015, 23(1): 13-17. |
| [6] |
王伯周, 翟连杰, 廉鹏, 等. 3, 3'-二(氟偕二硝基)二呋咱基醚(FOF-13)新法合成[J].
含能材料, 2014, 22(6): 884-886. WANG Bo-zhou, ZHAl Lian-jie, LlAN Peng, et al. A novel synthesis of 3, 3'-bis(fluorodinitromethyl)difurazanyl ether(FOF-13)[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(6): 884-886. DOI:10.11943/j.issn.1006-9941.2014.06.033 |
| [7] |
付小龙, 樊学忠, 毕福强, 等. 硝基呋咱/CMDB推进剂能量特性[J].
含能材料, 2014, 22(6): 852-856. FU Xiao-long, FAN Xue-zhong, Bl Fu-qiang, et al. Energy characteristics of CMDB propellants with Nitrofurazan compounds[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2014, 22(6): 852-856. DOI:10.11943/j.issn.1006-9941.2014.06.027 |
| [8] |
Luk'yanov O A, Pokhvisneva G V, Ternikova T V, et al. Aliphatic α-nitroalkyl-ONN-azoxy compounds and their derivatives[J].
Russian Chemical Bulletin, 2009, 58(10): 2063-2069. DOI:10.1007/s11172-009-0283-0 |
| [9] |
Luk'yanov O A, Pokhvisneva G V, Ternikova T V, et al. α-Nitroalkyl-ONN-azoxyfurazanes and some of their derivatives[J].
Russian Chemical Bulletin, 2011, 60(8): 1703-1711. DOI:10.1007/s11172-011-0254-0 |
| [10] |
Luk'yanov O A, Pokhvisneva G V, Ternikova T V, et al. 3, 4-Bis (α-nitroalkyl-ONN-azoxy) furazans and some of their derivatives[J].
Russian Chemical Bulletin, 2012, 61(2): 360-365. DOI:10.1007/s11172-012-0050-5 |
| [11] |
Luk'yanov O A, Pokhvisneva G V, Ternikova T V. Bis (nitro-and polynitromethyl-ONN-azoxy) azoxyfurazans and some of their derivatives[J].
Russian Chemical Bulletin, 2012, 61(9): 1783-1786. DOI:10.1007/s11172-012-0245-9 |
| [12] |
Luk'yanov O A, Parakhin V V, Pokhvisneva G V, et al. 3-Amino-4-(α-nitroalkyl-ONN-azoxy) furazans and some of their derivatives[J].
Russian Chemical Bulletin, 2012, 61(2): 355-359. DOI:10.1007/s11172-012-0049-y |
| [13] |
田均均, 张庆华, 李金山. 含能分子合成最新进展[J].
含能材料, 2016, 24(1): 1-9. TIAN Jun-jun, ZHANG Qing-hua, LI Jin-shan. Recent advances in energetic molecule synthesis[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2016, 24(1): 1-9. DOI:10.11943/j.issn.1006-9941.2016.01.001 |
| [14] |
Becke A D. Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].
Journal of Chemistry Physics, 1993, 98(7): 5648-5652. DOI:10.1063/1.464913 |
| [15] |
Lu T, Chen F. Multiwfn:a multifunctional wavefunction analyzer[J].
J Comput Chem, 2002, 33: 580-592. |
| [16] |
林梦海.
量子化学计算方法与应用[M]. 北京: 科学出版社, 2004.
LIN Meng-hai. Computational methods and applications of quantum chemistry[M]. Beijing: Science Press, 2004. |
| [17] |
Rice B M, Byrd E F C. Evaluation of electrostatic descriptors for predicting crystalline density[J].
J Comput Chem, 2013, 34: 2146-2151. DOI:10.1002/jcc.23369 |
| [18] |
Larry A.Curtiss, Krishnan Raghavachari, Paul C.Redfern, et al. Gaussian-3 G3 theory for molecules containingfirst and second-row atoms[J].
J Chem Phys, 1998, 109(18): 7764-7776. DOI:10.1063/1.477422 |
| [19] |
Politzer P, Murray J S, Grice M E, et al. Calculation of heats of sublimation and solid phase heats of formation[J].
Mol Phys, 1997, 91: 923-928. DOI:10.1080/002689797171030 |
| [20] |
Kamlet M J, Jacobs S J. Chemistry of detonationI.a simple method for calculating detonation properties of CHNO explosives[J].
Journal of Chemical Physics, 1968, 48(1): 23-25. DOI:10.1063/1.1667908 |
| [21] |
Pospísil M, Vavra P, Concha M C, Murray J S, Politzer P. A possible crystal volume factor in the impact sensitivities of some energetic compounds[J].
J Mol Model, 2010, 16: 895-901. DOI:10.1007/s00894-009-0587-x |
| [22] |
Hammerl A, Klapötke T M., Noeth H, et al. Synthesis, structure, molecular orbital and valence bond calculations for tetrazole azide, CHN7[J].
Propellants, Explosives, Pyrotechnics, 2003, 28: 165-173. DOI:10.1002/(ISSN)1521-4087 |
| [23] |
Jiaheng Zhang, Jean'ne M. Shreeve.3, 3'-Dinitroamino-4, 4'-azoxyfurazan and Its derivatives an assembly of diverse N-O building blocks for high-performance energetic materials[J].
J Am Chem Soc, 2014, 136(11): 4437-4445. DOI:10.1021/ja501176q |
| [24] |
肖鹤鸣, 许晓娟, 邱玲.
高能量密度材料的理论设计[M]. 北京: 科学出版社, 2008.
XIAO He-ming, XU Xiao-uan, QIU Lin. Theoretical design for high energetic materials[M]. Beijing: Science Press, 2008. |
The effect of nitroazoxy, trinitromethylazoxy and fluorodinitromethylazoxy on the properties of disubstituted furazan, azofurazan, azoxyfurazan and furazan ether were studied theoretically and comparatively. Based on the calculated results of 12 furazan energetic derivatives, considering that 3, 3′-bis(fluorodinitromethylazoxy)-4, 4′-azoxyfurazan is considered to be a high energy density compound.