




2. 北京故宫博物院文保科技部, 北京 100080
2. Conservation Technology Department, the Palace Museum, Beijing 100080, China
高氮含能化合物是近年来发展起来的并具有良好应用前景的高能量密度材料 (HEDM), 它具有很高的正生成焓, 感度较低, 热稳定性好, 闪点温度高, 且分子结构中的高氮低碳氢含量使其具有较高的密度, 也更容易达到氧平衡[1-2]。四嗪类高氮化合物分子结构中含有较多的N—N和C—N键, 四嗪环的含氮量高达68.3%, 具有能量高、热稳定性好、特征信号低、燃烧无残渣及无污染等优点, 广泛应用于推进剂、新型高能钝感炸药和烟火剂等含能材料领域[3-4]。而稠环含能化合物一般都具有较高的密度和能量, 是目前高能量密度材料重要研究方向之一[5-6]。3-肼基-6-(3, 5-二甲基吡唑)-s-四嗪可以发生成环反应形成稠环类四嗪含能化合物[7], 如s-四嗪并三唑的衍生物1, 2, 4-三唑[4, 3-b]并s-四嗪 (TTZ)、6-氨基-1, 2, 4-三唑[4, 3-b]并s-四嗪 (ATZ) 和3, 6-二氨基-1, 2, 4-三唑[4, 3-b]并s-四嗪 (AATZ)。其后, 研究者合成了许多稠环类四嗪衍生物[8-14], 但有关该类化合物的研究大多集中在该类物质的制备及机理方面的研究, 而有关性能研究相对较少。
基于此, 本研究选取了一系列五元氮杂环与母体1, 2, 4, 5-四嗪连接组成稠环化合物, 设计了14种1, 2, 4, 5-四嗪衍生物, 在wB97/6-31+G**水平下获得此类化合物的稳定构型, 在此基础上计算了其生成焓及爆轰性能, 从理论上研究五元氮杂环作为取代基构成的稠环化合物对1, 2, 4, 5-四嗪的影响, 考察性能与结构之间的对应关系。
2 计算方法运用DFT方法, 在B3LYP/(6-31G*, 6-311G*, 6-31+G*, 6-31G**, 6-311G**, 6-31+G**, 6-311++G**, cc-pVDZ, cc-pVTZ) 水平下计算分析1, 2, 4, 5-四嗪、3, 6-二氨基-1, 2, 4, 5-四嗪 (DAT)、3, 6-二肼基-1, 2, 4, 5-四嗪 (DHT) 和3, 6-二叠氮基-1, 2, 4, 5-四嗪 (DIAT)4种化合物的生成焓, 通过与实验值[15]进行对比拟合, 在B3LYP/6-31+G**水平下计算的结果与实验值线性相关性最好, 达到0.9863, 因此选择基组6-31+G**和不同方法 (B3PW91, M05, M05-2X, M06, M06-2X, wB97) 组合计算上述4种化合物生成焓, 通过与实验值进行对比拟合, 在wB97/6-31+G**水平下计算的结果与实验值线性相关性最好, 达到0.9896, 因此在wB97/6-31+G**水平下对所设计的14种1, 2, 4, 5-四嗪衍生物的几何结构进行全优化, 经振动频率分析表明优化构型为势能面上极小点 (无虚频), 得到的热力学数据采用原子化方案 (atomization scheme)[16-20]预测目标化合物的标准生成焓。具体方法是将分子分解为原子:
| $ {{\rm{C}}_a}{{\rm{H}}_b}{{\rm{O}}_c}{{\rm{N}}_d}\left( {\rm{g}} \right) \to a{\rm{C}}\left( {\rm{g}} \right) + b{\rm{H}}\left( {\rm{g}} \right) + c{\rm{O}}\left( {\rm{g}} \right) + d{\rm{N}}\left( {\rm{g}} \right) $ | (1) |
则该反应在298K时的标准反应焓ΔH298由下式计算:
| $ \begin{array}{l} \Delta {H_{298}} = \Sigma \Delta {H_{\rm{f}}}_{{\rm{, P}}}-\Sigma \Delta {H_{{\rm{f, R}}}}\\ = a\Delta {H_{\rm{f}}}_{{\rm{, C}}} + b\Delta {H_{{\rm{f, H}}}} + c\Delta {H_{{\rm{f, O}}}} + d\Delta {H_{{\rm{f, N}}}}-\Delta {H_{{\rm{f, }}{{\rm{C}}_a}{{\rm{H}}_b}{{\rm{O}}_c}{{\rm{N}}_d}}} \end{array} $ | (2) |
式中, ΔHf, R和ΔHf, p分别表示反应物和生成物在298 K的标准生成焓, kJ·mol-1; ΔHf, C、ΔHf, H、ΔHf, O和ΔHf, N分别为原子C、H、O和N在298 K的标准生成焓, 可从手册[21]中查得; ΔHf, CaHbOcNd为分子CaHbOcNd在298 K的标准生成焓, 为待求项。同时存在下列关系式:
| $ \begin{array}{l} \Delta {H_{298}} = \Delta {E_{298}} + \Delta (pV)\\ = \Delta {E_0} + \Delta {E_{{\rm{ZPE}}}} + \Delta {E_{\rm{T}}} + \Delta nRT\\ = {E_{0, {\rm{C}}}} + {E_{0, {\rm{H}}}} + {E_{0, {\rm{O}}}} + {E_{0, {\rm{N}}}}-{E_{0, {{\rm{C}}_a}{{\rm{H}}_b}{{\rm{O}}_c}{{\rm{N}}_d}}}-\\ {E_{{\rm{ZPE, }}{{\rm{C}}_a}{{\rm{H}}_b}{{\rm{O}}_c}{{\rm{N}}_d}}}-\Delta {E_{{\rm{T}}, {{\rm{C}}_a}{{\rm{H}}_b}{{\rm{O}}_c}{{\rm{N}}_d}}} + \Delta nRT \end{array} $ | (3) |
式中, E0, C、E0, H、E0, O、E0, N和E0, CaHbOcNd分别为wB97/6-31+G**水平下计算得到的原子C、H、O、N和分子CaHbOcNd在0 K的总能量, a.u.; EZPE, CaHbOcNd和ΔET, CaHbOcNd分别为分子CaHbOcNd的零点能和热校正值, a.u., 可从振动分析获得的热力学数据得到, 对原子而言, EZPE和ΔET项均为0; Δn表示气体产物和反应物的物质的量之差, mol; R是气体常数, 8.314 J·mol-1·K-1; T表示绝对温度, K。综合上式, 化合物CaHbOcNd在298 K的标准生成焓ΔHf, CaHbOcNd即可求得。
运用半经验K-J方程[22-25]估算其爆速 (D)、爆压 (p) 值:
| $ D = {\mathit{\Phi} ^{0.5}}(1.011 + 1.312\rho ) $ | (4) |
| $ p = 1.558{\mathit{\Phi}} {\rho ^2} $ | (5) |
其中
| $ {\mathit{\Phi}} = N\bar M{^{0.5}}{Q^{0.5}} $ | (6) |
式中, p为爆压, GPa; D为爆速, km·s-1; ρ为密度, g·cm-3; Φ为炸药的特性值, N为每克炸药气体爆轰产物摩尔数, mol·g-1; M为气体产物的平均摩尔质量, g·mol-1; Q为每克炸药的爆轰化学能, kJ·g-1; 结合原子化方案所得的生成焓, 参照文献[18]的方法求得N, M和Q, 具体方法如表 1所示。
| 表 1 CaHbOcNd中的参数N、M和Q的计算方法 Tab.1 The calculated methods of the parameters N、M and Q of CaHbOcNd |
研究中所有化合物密度均采用摩尔体积法 (ρ=M/Vm) 计算得到, 其中M为化合物的摩尔质量, Vm为化合物的摩尔体积, 是在稳定构型下, 基于0.001 e·bohr-3等电子密度面所包围的体积空间, 用Monte-Carlo方法对每一个优化的稳定构型进行了100次单点计算取其平均值求得。所有计算使用Gaussian09 [26]量子化学软件包在wB97/6-31+G**水平下完成。
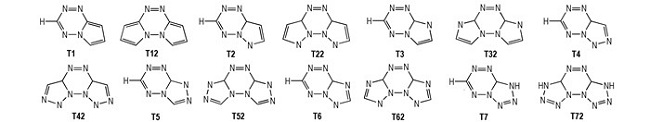
3 结果与讨论 3.1 几何结构在wB97/6-31+G**水平下对1, 2, 4, 5-四嗪衍生物进行几何结构全优化, 图 1列出了其优化结构图, 标注了部分优化几何参数 (键长、键角)。计算结果表明, 与1, 2, 4, 5-四嗪相比四嗪环上N (1)—N (2) 键长均缩短, 除了T2靠近五元环一侧的N (4)—N (5) 键长, 其余化合物的键长均增加, 双环取代更明显; 单环取代四嗪衍生物C (3)—N (4) 键长均缩短, 其他C—N键长均增加, 双环取代四嗪衍生物C—N键长均增加, 且大部分五元环的C—N键长小于未成五元环的; 同分异构体化合物中, 五元环上N原子位置不同, 使四嗪环的上的键长和键角不同, 但数值非常接近, 因此N原子位置对四嗪环的键长和键角影响较小; 除了衍生物T1和T12, 其余化合物均不共面, 说明随着N原子数量的增加, 稠环类四嗪衍生物的共面性反而降低。

|
图 1 1, 2, 4, 5-四嗪衍生物的分子优化结构图及部分键长 (nm) 和键角 (°) Fig.1 Optimized molecular structures for the 1, 2, 4, 5-tetrazine derivatives along with their selected bond lengths (nm) and bond angles (°) |
在wB97/6-31+G**水平下采用原子化方案估算稠环四嗪衍生物及传统含能材料RDX和HMX的生成焓, 表 2列出了目标化合物的总能量 (E0)、零点能 (EZPE)、N原子数、温度校正值 (HT) 及生成焓 (ΔHf)。
| 表 2 均四嗪衍生物及HMX和RDX的总能量、零点能、温度校正值和生成焓 Tab.2 Calculated E0, EZPE, HT and ΔHf of s-tetrazine derivatives together with HMX and RDX |
计算结果表明, 所有1, 2, 4, 5-四嗪衍生物生成焓均大于传统含能材料RDX和HMX, 具有高正生成焓, 其中最高的生成焓值为1122.53 kJ·mol-1。所有化合物的生成焓均比未取代的1, 2, 4, 5-四嗪生成焓高, 双环取代四嗪衍生物生成焓普遍大于相应单环取代的, 幅度为140~280 kJ·mol-1。单环取代中, 化合物T2和T3, T4、T5和T6分别互为同分异构体, T2的生成焓高于T3, T4的生成焓高于T5和T6, 是因为T2中的N—N键数量多于T3, T4中的N—N键数量多于T5和T6, 双环取代 (T22、T42) 和单环取代一致, 结果表明, N—N键有助于增加1, 2, 4, 5-四嗪衍生物的生成焓。
选取相同N原子数中生成焓最大的化合物, 对其总能量 (E0) 和生成焓 (ΔHf) 与N原子数的关系作线性拟合, 由图 2和图 3可知, 不论是单环取代还是双环取代, 随着N原子数的增加, 分子总能量逐渐降低, 且具有很好的线性关系, 生成焓则逐渐增大, 表明N原子数的增加有助于提升1, 2, 4, 5-四嗪衍生物的生成焓。综上所述, N—N键及N原子数的增加在提升1, 2, 4, 5-四嗪衍生物的生成焓方面起了重要的作用。

|
图 2 总能量 (E0) 与N原子数的关系 Fig.2 The relationship of E0 and N atom numbers |

|
图 3 生成焓 (ΔHf) 与N原子数的关系 Fig.3 The relationship of ΔHf and N atom numbers |
分子轨道理论表明, 化合物的稳定性与其分子轨道能量有关, 最高占据轨道能量 (EHOMO) 越低, 最低空轨道能量 (ELUMO) 越高, 则其分子轨道能级差 (ΔELUMO-HOMO=ELUMO-EHOMO) 越大, 化合物就越稳定。运用量子化学的方法计算了1, 2, 4, 5-四嗪衍生物的EHOMO及ELUMO, 进一步分析得到ΔELUMO-HOMO, 列于表 3。计算结果表明, 单环取代衍生物的分子轨道能级差与T比较均减小, 双环取代衍生物除了T12和T32, 其它分子轨道能级差与T比较均增大, T62的分子轨道能级差在所有体系中较高, 使电子跃迁几率降低, 预示其反应活性最低, 最稳定。
| 表 3 目标化合物的前线轨道能量 Tab.3 Calculated EHOMO, ELUMO和ΔELUMO-HOMO of the title compound |
爆速和爆压是研究爆轰性能的两个较为重要的参数, 本文在wB97/6-31+G**水平下预测了1, 2, 4, 5-四嗪衍生物及HMX和RDX的爆速 (D) 和爆压 (p), 如表 4所示。无论是单环取代还是双环取代的衍生物, D和p都是随着体系中N原子数的增加而增加; 除了单环取代的T1、T2、T3及双环取代的T12、T22、T32, 其他衍生物的密度、爆速和爆压均高于未取代的T; 带有同一种环单取代和双取代, 对D和p的影响较小, 且有些单环取代衍生物的D和p略偏高, 如单环取代化合物T1、T2和T3比相应双环取代的T12、T22、T32高, 说明稠环四嗪衍生物D和p与所含N原子数关系较大, 与环的个数关系较小。化合物T7和T72的D接近于传统含能材料RDX, p略低于RDX, ρ则远远小于RDX, 从能量角度来看, 提高四嗪衍生物的密度, 其爆轰性能也将提高, 所以T7和T72可以作为潜在的含能材料。
| 表 4 目标化合物及RDX和HMX的摩尔质量、平均摩尔体积、理论密度、爆热、爆速和爆压 Tab.4 Predicted M, V, ρ, Q, D and p of the title molecules together with RDX and HMX |
1, 2, 4, 5-四嗪衍生物的D和p与N原子数的线性拟合关系如图 4所示, 图 4a与图 4c为单环取代四嗪衍生物的D和p与N原子数的线性关系, 图 4b与图 4d为双环取代四嗪衍生物的D和p与N原子数的线性关系。从图 4可以看出, 无论是单环取代还是双环取代, 1, 2, 4, 5-四嗪衍生物的D和p与N原子数均有很好的线性关系, 相关系数r分别为0.987、0.998(单取代D、p) 和0.988、0.996(双取代D、p)。

|
图 4 爆速 (D) 及爆压 (p) 与氮原子数目的关系 Fig.4 The relationship of D and p with N atom numbers |
运用量子化学计算的方法, 以分子统计热力学为基础, 计算了稠环类1, 2, 4, 5-四嗪衍生物在200~800K的热力学性质, 即标准摩尔热容 (Cp, m)、标准摩尔熵 (Sm) 和标准摩尔焓 (Hm), 列于表 5。由表 5可以看出, Cp, m、Sm及Hm均随T的升高而增加, 其中Cp, m和Sm增大的比例均随着T的升高而逐步减小, 而Hm增大的比例则随着T的升高而逐步增大。在T较低时, 分子的转动及平动对Cp, m、Sm和Hm贡献相对较大; 但是随着温度升高到一定程度后, 分子的振动增强, 对Cp, m、Sm和Hm贡献大, 而导致Cp, m、Sm和Hm值增加。在同一T下, 随着N原子数的增加, Cp, m、Sm及Hm与温度之间没有线性的变化; 同时, 双取代环的Cp, m、Sm和Hm明显大于相应的单取代环的, 说明取代环的增加有利于这些热力学函数值的增加。
| 表 5 目标化合物200~800 K时的Cp, m、Sm和Hm Tab.5 Calculated Cp, m, Sm and Hm at 200~800 K for the title molecules |
利用wB97/6-31+G**方法对稠环类1, 2, 4, 5-四嗪衍生物的几何结构、前线轨道能量、生成焓、爆轰性能及热力学性质进行计算研究。
(1) 计算结果表明, 双环取代四嗪衍生物生成焓普遍大于相应单环取代的, N—N键及N原子数的增加有助于提升1, 2, 4, 5-四嗪衍生物的生成焓。
(2) 爆轰性能结果表明, 稠环四嗪衍生物D和p主要与所含N原子数有关, 与环的个数关系较小, 且D和p与N原子数均有良好的一次线性相关关系。
(3) 热容Cp, m、熵Sm及焓Hm均随着T的升高而增加, Cp, m和Sm增大的比例均随着T的升高而逐步减小, 而Hm增大的比例则随着T的升高而逐步增大。
(4) 化合物T7和T72的爆速接近于传统含能材料RDX, 爆压略低于RDX, 可以作为备选的HEDM。
致谢: 感谢临沂大学化学化工学院夏其英教授在Gaussian09计算中提供的帮助。| [1] | Fair H D, Walker R F. EnergeticMaterials[M]. Springer Berlin, 2014. |
| [2] |
张兴高, 朱慧, 张炜, 等. 高氮化合物在含能材料中的应用研究进展[J].
含能材料, 2004, 12: 48-53. ZHANG Xing-gao, ZHU Hui, ZHANG Wei, et al. Application development of poly-nitrogen compounds in energetic materials[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2004, 12: 48-53. DOI:10.3969/j.issn.1006-9941.2004.01.015 |
| [3] |
周阳, 龙新平, 王欣, 等. 高氮含能化合物的研究新进展[J].
含能材料, 2006, 14(4): 315-320. ZHOU Yang, LONG Xin-ping, WANG Xin, et al. Review on high-nitrogen energetic materials[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2006, 14(4): 315-320. |
| [4] |
雷永鹏, 徐松林, 阳世清. 高氮含能化合物应用研究新进展[J].
化学推进剂与高分子材料, 2007, 5(3): 1-14. LEI Yong-peng, XU Song-lin, YANG Shi-qing. New research progress in application of high-nitrogen energetic compound[J]. Chemical Propellants & Polymeric Materials, 2007, 5(3): 1-14. |
| [5] |
霍欢, 王伯周, 廉鹏, 等. 三种含能稠环化合物合成与表征研究[C]//第二届全国危险物质与安全应急技术研讨会论文集, 成都, 2013: 41-45.
HUO Huan, WANG Bo-zhou, LIAN Peng, et al. Synthesis and characterization of 3 energetic fused ring compounds[C]//Proceedings of the 2nd national seminar of hazardos materials & security emergency technology, Chengdu, 2013: 41-45. |
| [6] |
田均均, 张庆华, 李金山. 含能分子合成最新进展[J].
含能材料, 2016, 24(1): 1-9. TIAN Jun-jun, ZHANG Ling-hua, LI Jin-shan. Recent advances in energetic molecule synthesis[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2016, 24(1): 1-9. DOI:10.11943/j.issn.1006-9941.2016.01.001 |
| [7] | Chavez D E, Hiskey M A. Synthesis of the bi-heterocyclic parent ring system 1, 2, 4-triazolo[4, 3-b][1, 2, 4, 5] tetrazine and some 3, 6-disubstituted derivatives[J]. Journal of Heterocyclic Chemistry, 1998, 35(6): 1329-1332. DOI:10.1002/jhet.v35:6 |
| [8] | Palysaeva N V, Kumpan K P, Struchkova M I, et al. A direct approach to a 6-hetarylamino[1, 2, 4] triazolo[4, 3-b][1, 2, 4, 5] tetrazine library[J]. Organic Letters, 2014, 16: 406-409. DOI:10.1021/ol403308h |
| [9] | Tolshchina S G, VyakhirevaA G, Ignatenko N K, et al. Cyclization of (1, 2, 4, 5-tetrazin-3-yl) hydrazones to 3, 7-dihydro-1, 2, 4-triazolo[4, 3-b]-1, 2, 4, 5-tetrazines[J]. Russian Chemical Bulletin (International Edition), 2009, 58(6): 1281-1290. DOI:10.1007/s11172-009-0168-2 |
| [10] | Myers T W, Bjorgaard J A, Brown K E, et al. Energetic chromophores: low-energy laser initiation in explosive Fe (Ⅱ) tetrazine complexes[J]. Journal of the American Chemical Society, 2016, 138: 4685-4692. DOI:10.1021/jacs.6b02155 |
| [11] | Sheremetev A B, Palysaeva N V, Suponitskii K Y, et al. Synthesis of[1, 2, 4] triazolo[4, 3-b]-s-tetrazines with incorporated furazan ring[J]. Russian Chemical Bulletin (International Edition), 2012, 61(1): 121-130. DOI:10.1007/s11172-012-0017-6 |
| [12] | Novák Z, Csámpai A, Kotschy A. Synthesis and alkylation of some[1, 2, 4] triazolo[4, 3-b]tetrazines[J]. Arkivoc, 2000(ⅲ): 259-265. |
| [13] | Rudakov G F, Borodavina A D, Efimova P A, et al. Synthesis of energetic materials based on 1, 2, 4-triazolo[4, 3-b]-1, 2, 4, 5-tetrazine[C]//10th International Autumn Seminar on Propellants, Explosives & Pyrotechnics, Chengdu, 2013. |
| [14] | Ganebnykh I N, Tolshchina S G, Ishmetova R I, et al. Unusual expansion of the 1, 2, 4, 5-tetrazine ring in[1, 2, 4] triazolo[4, 3-b]-[1, 2, 4, 5] tetrazines leading to[1, 2, 4, 6] tetrazepine systems[J]. European Journal of Organic Chemistry, 2011, 2011(12): 2309-2318. DOI:10.1002/ejoc.v2011.12 |
| [15] | WEI Tao, ZHU Wei-hua, ZHANG Xiao-wen, et al. Molecular design of 1, 2, 4, 5-tetrazine-based high-energy materials[J]. J Phy Chem A, 2009, 113: 9404-9412. DOI:10.1021/jp902295v |
| [16] |
邱丽美, 贡雪东, 郑剑, 等. 由原子化反应法求算高能化合物的生成热[J].
含能材料, 2008, 16(6): 647-668. QIU Li-mei, GONG Xue-dong, ZHENG Jian, et al. Heats of formation for energetic compounds calculated using atomization reactions[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2008, 16(6): 647-668. |
| [17] | ZHOU Yang, LONG Xin-ping, SHU Yuan-jie. Theoretical studies on the heats of formation, densities, and detonation properties of substituted s-tetrazine compounds[J]. Journal of Molecular Modeling, 2010, 16: 1021-1027. DOI:10.1007/s00894-009-0605-z |
| [18] | PAN Yong, ZHU Wei-hua, XIAO He-ming. Theoretical studies on the structures, heats of formation, energetic properties and pyrolysis mechanisms of nitrogen-rich difurazano[3, 4-b:3′, 4′-e]piperazine derivatives and their analogues[J]. Structural Chemistry, 2013, 24: 1071-1087. DOI:10.1007/s11224-012-0133-9 |
| [19] | WANG Fang, WANG Gui-xiang, DU Hong-chen, et al. Theoretical Studies on the Heats of Formation, Detonation properties, and pyrolysis mechanisms of energetic cyclic nitramines[J]. Journal of Physical Chemistry A, 2011, 115: 13858-13864. DOI:10.1021/jp2047536 |
| [20] | LI Xiao-hong, ZHANG Rui-zhou, ZHANG Xian-zhou. Theoretical studies of 1, 2, 4, 5-tetrazine-based energetic nitrogen-rich compounds[J]. Structural Chemistry, 2013, 24: 393-400. DOI:10.1007/s11224-012-0089-9 |
| [21] | David R L. CRC Handbook of Chemistry and Physics[M]. LLC: CRC Press, 2005: 5-1, 2. |
| [22] | Kamlet M J, Jacobs S J. Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives[J]. Journal of Chemical Physics, 1968, 48(1): 23-25. DOI:10.1063/1.1667908 |
| [23] |
王桂香, 肖鹤鸣, 居学海, 等. 含能材料的密度、爆速、爆压和静电感度的理论研究[J].
化学学报, 2007, 65(6): 517-524. WANG Gui-xiang, XIAO He-ming, JU Xue-hai, et al. Theoretical studies on densities, detonation velocities and pressures and electric spark sensitivities of energetic materials[J]. Acta Chimica Sinica, 2007, 65(6): 517-524. |
| [24] |
李志敏, 严英俊, 冀慧莹, 等. 苦味酸含能离子盐的结构、生成热及爆炸性能理论研究[J].
火炸药学报, 2009, 32(6): 6-10. LI Zhi-min, YAN Ying-jun, JI Hui-ying, et al. Theoretical study on structure, heat of formation and detonation properties of picrate-based energetic ionic salts[J]. Chinese Journal of Explosives & Propellants, 2009, 32(6): 6-10. |
| [25] |
张熙和, 云主慧.
爆炸化学[M]. 北京: 国防工业出版社, 1989: 222-241.
ZHANG Xi-he, YUN Zhu-hui. Explosion chemistry[M]. Beijing: National Defence Industry Press, 1989: 222-241. |
| [26] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A.02[CP]. Gaussian, Inc.: Wallingford, CT, 2009. |
| [27] | Talawar M B, Sivabalan B, Mukundan T, et al. Environmentally compatible next generation of 'green' energetic materials (GEMs)[J]. Journal of Hazardous Materials, 2009, 161(2-3): 589-607. DOI:10.1016/j.jhazmat.2008.04.011 |
The optimized geometries and enthalpies of formation (ΔHf) of 14 kinds of 1, 2, 4, 5-tetrazine polycyclic ring derivatives were studied using density functional theory method. The detonation performances were estimated by Kamlet-Jacobs equation.