



呋咱类含能化合物由于具有高能量密度、高标准生成焓、高氮含量等优点, 是含能材料领域备受关注的研究方向之一[1-3]。其中3, 4-双(4′-硝基呋咱-3′-基)氧化呋咱(DNTF)集呋咱、氧化呋咱及硝基于一体,在众多呋咱化合物中最为典型,综合性能优于奥克托今(HMX),特别是其熔点低,可用作熔铸炸药中的液相载体,已成为新一代高能量密度材料[4-6],具有广阔的应用前景。在DNTF结构表征方面已经获得完整的红外、质谱数据,然而,核磁共振(NMR)表征研究却不够深入,由于DNTF结构中不含氢,仅由对NMR不灵敏核的C、N、O三种元素组成,尽管研究人员获得了其碳谱和氮谱数据[5-7],但因缺乏相关的经验数据及理论支撑,无法直接对NMR信号准确归属。另外,因缺乏灵敏有效的NMR相关机制,常规的二维NMR(大多为含氢化合物设计)也无能为力,为该类化合物核磁表征研究带来极大困难。作为NMR实验的有益补充,NMR的理论计算也受到越来越多的关注,在指认与解释图谱方面发挥了重要的作用[8-11]。马海霞等[12]通过理论计算值与实验值对比,对DNTF的核磁信号进行归属,但是仅计算了碳谱化学位移,且因实验数据不全(获得了6个碳信号中的4个信号),归属出现错误。
本实验则详细研究了DNTF在不同氘代试剂溶液中的碳谱与氮谱,采用二维碳碳相关稀核双量子转移实验(Incredible Natural Abundance Double Quantum Transfer Experiment,INADEQUATE) [13]完成了碳信号的全归属。同时,利用量子化学密度泛函理论(Density Functional Theory, DFT)[14-16]对DNTF的结构和13C、15N NMR化学位移进行了理论研究,进一步从理论角度解释了DNTF的分子结构和NMR化学位移之间的关系,为呋咱类化合物NMR研究提供技术和理论参考。
2 实验 2.1 主要仪器与试剂试剂:DNTF,纯度≥99.5%,西安近代化学研究所;氘代二甲基亚砜(DMSO-d6,99.8%)、氘代丙酮(Acetone-d6,99.8%)、氘代氯仿(CDCl3,99.8%),美国CIL公司。
仪器:瑞士Bruker公司AV500型(500MHz)超导核磁共振仪。
2.2 仪器条件一维实验:采用正向检测探头(BBO多核宽带探头), Φ5 mm样品管, 1H NMR观测频率为500.13 MHz, 13C NMR的观测频率为125.77MHz, 15N NMR的观测频率为50.69 MHz。1H、13C NMR以TMS为内标(δ 0),15N NMR以硝基甲烷(CH3NO2)为外标(δN 0);二维INADEQUATE实验:采样点阵t2×t1=8192×128, FT变换数据点阵F2×F1=8192×1024,累加次数ns=256,脉冲延迟时间d1=5 s, 混合时间d2=7.6 ms。
2.3 计算方法运用Gaussian 09程序,以密度泛函理论的B3LYP[17]方法在6-311+G(2d, p)基组水平下对DNTF分子进行几何全优化。对优化后的构型进行振动分析,无虚频,表明其对应自势能面上的极小点。用GIAO方法分别在B3LYP/6-311G、B3LYP/6-311G(d)、B3LYP/6-311G(2d, p)、B3LYP/6-311+G(2d, p)水平上计算了DNTF的核磁共振碳谱与氮谱。
3 结果与讨论 3.1 不同氘代试剂溶液中DNTF的谱图特征研究了DNTF在氘代氯仿、氘代丙酮和氘代二甲基亚砜溶液中的谱图特征,如图 1和图 2所示。DNTF在3种溶剂中的碳谱和氮谱化学位移值列于表 1。碳谱中,丙酮与氯仿溶液中在δ160附近信号明显呈多重峰,这可能是由于14N核偶合所致,但只能发现5条谱线,与结构中的6个碳原子数目不相符。而在DMSO溶液中可明显观察到6条谱线,与分子结构一致,且碳谱位移在δ160.43、δ160.24出现明显可区分信号,因此,推测在丙酮、氯仿溶液中,δ160附近的信号应为2条谱线叠加而难以区分,且由于弛豫时间长而信号较弱。文献[12] NMR观测到的实验结果见表 1,结果与本实验丙酮溶液中的较强信号基本一致,本实验还观测到δ160附近较弱的2个信号。另外,DNTF在氯仿溶液中溶解度较差,所以其碳谱信噪比较差,氮谱则难以检出。丙酮与二甲基亚砜中可观测到8条尖锐的氮谱谱峰,与结构中的8个氮原子一致。由于溶剂效应,样品在3种溶剂中的化学位移略有差别,但基本一致。但因化学环境接近而不易归属各个碳与氮原子,需要进一步从实验与理论研究中获得准确归属。

|
图 1 DNTF在氘代二甲基亚砜(A)、氘代丙酮(B)和氘代氯仿(C)中的13C谱 Fig.1 13C NMR Spectra of DNTF in DMSO-d6(A), acetone-d6(B) and CDCl3(C) |

|
图 2 DNTF在氘代二甲基亚砜(A)、氘代丙酮(B)中的15N谱 Fig.2 15N NMR Spectra of DNTF in DMSO-d6(A) and acetone-d6(B) |
| 表 1 DNTF在不同氘代试剂中的13C, 15N NMR数据 Tab.1 13C and 15N NMR of DNTF in different solvent |
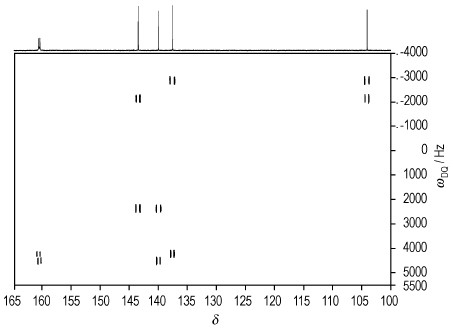
在氘代DMSO溶液中全面检测DNTF的13C NMR、15N NMR基础上,通过二维INADEQUATE实验技术进行了碳信号的全归属。DNTF分子中不含氢,所有的碳原子均为季碳,如图 3所示,故无法利用13C-1H相关信号对碳原子进行指配,因此,季碳的指配存在较大的困难,二维(2D)13C-13C INADEQUATE NMR实验可用于确定分子骨架中碳原子的连接顺序,即:在2D INADEQUATE谱图中,横轴为13C核的化学位移,纵轴为双量子频率,所有偶合的(或相邻的)一对13C核形成AX或AB自旋体系,在同一水平线上左右对称的处于准对角线的两侧。据此可以确定相邻的碳原子,进而连接出整个分子的碳原子骨架,是碳与碳相连及结构确认的最有效的手段,但通常因灵敏度太低而难以实现。DNTF在DMSO溶液中的溶解度较大,较高浓度可大幅提高INADEQUATE实验的灵敏度,因此,采用该技术可完成对DNTF碳骨架的确认。

|
图 3 DNTF结构 Fig.3 Structure of DNTF |
二维INADEQUATE实验(图 4)结果表明,DNTF分子中的6个不同化学位移的碳原子具有δ160.43→δ137.31→δ103.80→δ143.25→δ139.77→δ160.24的连接顺序。从结构分析可知,O(22)的存在使得C(9)与C(13)的化学环境出现较大的差异,而对C(2)、C(3)、C(14)、C(15)的影响较小,因此,δ103.80与δ143.25应为C(9)或C(13)的信号峰,由于N(10)→O(22)键的拉电子作用,使得氧化呋咱环中的电子云偏向于N(10),从而使得邻位碳原子C(9)的屏蔽增加而处在高场,因此,δ103.80为C(9),相应的,δ143.25为C(13),根据图 4及DNTF中碳链的连接顺序,可完成13C NMR的全归属,按照碳原子连接顺序其归属依次为:C(2)(δ160.43)、C(3)(δ137.31)、C(9)(δ103.80)、C(13)(δ143.25)、C(14)(δ139.77)、C(15)(δ160.24)。而对比文献[12]中仅有C(9)的信号归属正确,由于缺失的δ160附近2个碳信号引起其他碳信号的错误归属。

|
图 4 DNTF的13C-13C INADEQUATE图(DMSO-d6) Fig.4 13C-13C INADEQUATE spectrum of DNTF(DMSO-d6) |
传统的从头算方法与密度泛函理论已被广泛应用于表征电子作用。从头计算方法由于花费太大而只限在小分子体系中使用,而DFT方法能够廉价有效描述电子相互作用及交换,其中的B3LYP方法是NMR理论计算应用最广的一种方法。
采用密度泛函理论的B3LYP方法在6-311+G(2d, p)基组水平上对DNTF的几何结构进行了全优化,经振动频率分析,结果表明无虚频,说明该构型为势能面上的极小点,为相对稳定构型,其结构见图 5,其键长键角的计算结果见表 2。从表 2可知,DNTF稳定构型键长键角数据与文献[5]报道的晶体结构基本一致,说明该计算结果较为可靠。DNTF三个环处于不同平面,两个呋咱环与氧化呋咱环的夹角分别为-47.27°与47.60°。硝基上的氧原子与呋咱环不在同一平面,分别偏离呋咱环平面10.49°与6.81°。

|
图 5 DNTF优化结构 Fig.5 Optimized structure of DNTF |
| 表 2 The bond lengths and bond angles |
规范不变原子轨道(Gauge Independent Atomic Orbital, GIAO)[18-19]方法是目前公认的预测核磁共振化学位移较为准确的方法,已成功用于一些小分子及大中分子的NMR预测。在结构优化后,用GIAO方法,在不同基组水平上对DNTF分子中C、N原子的化学位移进行理论计算,因计算所得为绝对屏蔽值,其相对化学位移通过参比化合物(四甲基硅烷与硝基甲烷)的屏蔽值进行校准,其计算结果与实验结果见表 3和表 4。计算结果表明,B3LYP方法计算化学位移低估屏蔽而使计算结果偏大,随着计算基组的增大,与实验结果也越接近。对计算结果与实验数据进行线性回归,获得回归方程及相关系数列入表 3和表 4中。
| 表 3 DNTF的13C NMR实验结果与理论计算结果 Tab.3 Experimental and calculated chemical shifts of 13C NMR for DNTF |
| 表 4 DNTF的15N NMR实验结果与理论计算结果 Tab.4 Experimental and calculated chemical shifts of 15N NMR for DNTF |
从相关系数上看,计算结果与实验值吻合较好。碳谱计算尤为准确,小基组就可以获得较好的计算结果,最大误差小于10,经回归后,与实验结果相符程度非常高,可应用于核磁的谱线解释与归属。与之相比,氮谱计算更为复杂,误差也更大,但回归方程显示结果仍然较为理想,相关系数均高于0.99,对氮谱信号的归属具有参考价值。另外,氮谱计算结果显示在低频区计算结果与实验值符合较好,而对于呋咱环上的氮则相对较差,推测其原因为理论计算仅考虑单个分子上的电子屏蔽效应,事实上,由于氮灵敏度低,需要在高浓度下完成NMR测试,故需要考虑分子间的相互作用,从DNTF结构看,计算偏差大的几组氮原子均与氧原子距离接近,在浓度较大时,可能会发生相互作用,引起该几组氮的位移变化。
综上所述,DFT结合GIAO方法计算呋咱类含能化合物的核磁化学位移较为可靠,与实验结果一致,可应用于该类化合物的结构解析。
4 结论(1) DNTF的碳谱与氮谱在二甲基亚砜、丙酮、氯仿中的NMR实验结果基本一致,在二甲基亚砜中碳谱的分辨率最高,可获得完整的碳谱信息。
(2) 采用二维INADEQUATE实验获得了DNTF分子中碳原子之间的连接顺序,通过分析氧化呋咱环上配位氧原子的吸电子作用,确定了C(9)与C(13)的化学位移,完成了DNTF分子中碳信号的全归属。
(3) 在DNTF优化构型的基础上,以密度泛函理论的B3LYP方法结合GIAO方法计算获得了DNTF的碳谱和氮谱化学位移,与实验结果吻合较好,相关系数均大于0.99。
| [1] |
张叶高, 王伯周, 刘愆, 等. 5-[4-硝基呋咱基]-5H-[1, 2, 3]三唑并[4, 5-c][1, 2, 5]呋咱内盐(NOTO)合成与表征[J].
含能材料, 2010, 18(4): 383-386. ZHANG Ye-gao, WANG Bo-zhou, LIU Qian, et al. Synthesis of 5-(4-nitro-1, 2, 5-oxadiazol-3-yl)-5H-[1, 2, 3]triazolo[4, 5-c][1, 2, 5] oxadiazolium inner salt[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2010, 18(4): 383-386. |
| [2] |
李亚南, 张志忠, 周彦水, 等. 3, 4-二(吡嗪-2'-基)氧化呋咱的合成与表征[J].
火炸药学报, 2009, 32(6): 40-43. LI Ya-nan, ZHANG Zhi-zhong, ZHOU Yan-shui, et al. Synthesis and characterization of 3, 4-bis(pyrazine-2'-yl)furoxan[J]. Chinese Journal of Explosives & Propellants, 2009, 32(6): 40-43. |
| [3] |
薛云娜, 杨建明, 李春迎, 等. 3, 4-二苯基氧化呋咱的高效合成[J].
火炸药学报, 2010, 33(1): 34-36. XUE Yun-na, YANG Jian-ming, LI Chun-ying, et al. Synthesis of 3, 4-diphenylfuroxan with high efficiency[J]. Chinese Journal of Explosives & Propellants, 2010, 33(1): 34-36. |
| [4] |
胡焕性, 覃光明, 张志忠. 3, 4-二硝基呋咱基氧化呋咱炸药: CN02101092. 7[P], 2002.
HU Huan-xing, QIN Guang-ming, ZHANG Zhi-zhong. 3, 4-Dinitrofurazanfuroxan explosive:CN 02101092.7[P], 2002. |
| [5] |
周彦水, 张志忠, 李建康, 等. 3, 4-二硝基呋咱氧化呋咱的晶体结构[J].
火炸药学报, 2005, 28(2): 43-46. ZHOU Yan-shui, ZHANG Zhi-zhong, LI Jian-kang, et al. Crystal structure of 3, 4-dinitrofurazanofuroxan[J]. Chinese Journal of Explosives & Propellants, 2005, 28(2): 43-46. |
| [6] |
王军, 董海山, 黄奕刚, 等. 3, 4-二(硝基呋咱基)氧化呋咱的晶体结构研究[J].
含能材料, 2006, 14(5): 374-376. WANG Jun, DONG Hai-shan, HUANG Yi-gang, et al. Crystal structure of 3, 4-bis(nitrofurazano) furoxan[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2006, 14(5): 374-376. |
| [7] |
周彦水, 王伯周, 李建康, 等. 3, 4-双(4'-硝基呋咱-3'-基)氧化呋咱合成、表征与性能研究[J].
化学学报, 2012, 69(14): 1673-1680. ZHOU Yan-shui, WANG Bo-zhou, LI Jian-kang, et al. Study on synthesis, characterization and properties of 3, 4-bis(4'-nitrofurazano-3'-yl)furoxan[J]. Acta Chimica Sinica, 2012, 69(14): 1673-1680. |
| [8] |
刘兴艳, 廖显威, 陈国力, 等. 几种磺酰脲类除草剂的1H NMR谱的理论研究[J].
波谱学杂志, 2008, 25(2): 211-215. LIU Xing-yan, LIAO Xian-wei, CHEN Guo-li, et al. Theoretical calculation of 1H NMR spectra of sulfonylurea herbicides[J]. Chinese Journal of Magnetic Resonace, 2008, 25(2): 211-215. |
| [9] |
徐志广, 古国榜, 吴松平, 等. 2-丁基-四氢噻吩亚砜13C-NMR的理论研究[J].
结构化学, 2004, 23(10): 1183-1188. XU Zhi-guang, GU Guo-bang, WU Song-ping, et al. A theoretical study on 13C-NMR of 2-butyl-tetrahydrothiophene-1-oxide[J]. Chinese J Struct Chem, 2004, 23(10): 1183-1188. DOI:10.3969/j.issn.0254-5861.2004.10.017 |
| [10] |
陈海燕, 林翠梧, 陈光英, 等. Mycoepoxydiene核磁共振谱的理论研究[J].
化学研究, 2006, 17(4): 6-9. CHEN Hai-yan, LIN Cui-wu, CHEN Guang-ying, et al. Theoretical study on the NMR of Mycoepoxydiene[J]. Chemical Research, 2006, 17(4): 6-9. |
| [11] |
任洁, 朱华结. 计算化学在手性化合物结构分析中的应用[J].
高等学校化学学报, 2009, 30(10): 1907-1918. REN Jie, ZHU Hua-Jie. Application of computational chemistry in identification for chiral compounds[J]. Chemical Journal of Chinese Universities, 2009, 30(10): 1907-1918. DOI:10.3321/j.issn:0251-0790.2009.10.001 |
| [12] |
马海霞, 宋纪蓉, 肖鹤鸣, 等. 3, 4-二硝基呋咱基氧化呋咱(DNTF)的密度泛函理论研究[J].
火炸药学报, 2006, 29(3): 43-46. MA Hai-xia, SONG Ji-rong, XIAO He-ming, et al. Density functional theoretical investigation on 3, 4-dinitrofurazanfuroxan(DNTF)[J]. Chinese Journal of Explosives & Propellants, 2006, 29(3): 43-46. |
| [13] |
Lee S G. Carbon-13 two-dimensional INADEQUATE experiment of coprostane[J].
Bull Korean Chem Soc, 2001, 22(4): 429-431. |
| [14] |
李玉芳, 廖昕, 居学海, 等. 多叠氮基嗪异构化反应的密度泛函理论研究[J].
含能材料, 2010, 18(3): 241-246. LI Yu-fang, LIAO Xin, JU Xue-hai, et al. Density functional theory study on tautomerization of polyazido-azine[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2010, 18(3): 241-246. |
| [15] |
林秋汉, 李玉川, 祁才, 等. 6, 6'-二氨基氧化偶氮-1, 2, 4, 5-四嗪-1, 1', 5, 5'-四氧化物(DAATO5)的密度泛函理论[J].
火炸药学报, 2010, 33(3): 21-24. LIN Qiu-han, LI Yu-chuan, QI Cai, et al. Density functional theory of 6, 6'-diamino-oxidation of azo-1, 2, 4, 5-tetrazine-1, 1', 5, 5'-4-oxide compound[J]. Chinese Journal of Explosives & Propellants, 2010, 33(3): 21-24. |
| [16] |
胡银, 马海霞, 李军锋, 等. ATO的DFT研究、热力学性质及绝热至爆时间[J].
火炸药学报, 2009, 32(4): 18-21. HU Yin, MA Hai-xia, LI Jun-feng, et al. A density-functional theoretical investigation, thermodynamic properties and adiabatic time-to-explosion of ATO[J]. Chinese Journal of Explosives & Propellants, 2009, 32(4): 18-21. |
| [17] |
史秀锋, 孟瑞娟, 庞先勇, 等. C(24)团簇异构体的量子化学计算[J].
现代化工, 2009, 29(增刊): 347-350. SHI Xiu-feng, MENG Rui-juan, PANG Xian-yong, et al. Quantum chemical investigation of C(24) clusters isomers[J]. Modern Chemical Industry, 2009, 29(supple): 347-350. |
| [18] |
Knijn P J, van Bentum P J M, van Eck E R H, et al. A solid-state NMR and DFT study of compositional modulations in AlxGa1-xAs[J].
Phys Chem Chem Phys, 2010, 12: 11517-11535. DOI:10.1039/c003624b |
| [19] |
Li Y, Gao H, Zhang J, et al. Comparison of GIAO and CSGT for calculating 13C and 15N nuclear magnetic resonance chemical shifts of substituent neutral 5-aminotetrazole and 5-nitrotetrazole compounds[J].
Magn Reson Chem, 2012, 50: 16-21. DOI:10.1002/mrc.v50.1 |
In order to optimize the NMR assignment of 3, 4-dinitrofurazanfuroxan (DNTF), a combination of experimental NMR and computational GIAO-NMR techniques was utilized to distinguish the chemical shifts of 13C and 15N.