




2. 氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065
2. State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi′ an 710065, China
经过多年的发展,碳化学中关于碳骨架的构建,包括碳链增长、减短、异构、成环、开环等已形成了一系列成熟的理论和方法。而氮化学,自1772年分离出N2和1890年发现N3-以来,进展缓慢。近年来,大量的理论研究表明全氮化合物可能是潜在的高能量密度材料[1-4],氮化学研究也引起了广泛的兴趣,不仅合成出了具有划时代意义的N5+[5-6]和N5-[7-10]全氮离子,而且支撑该领域发展的氮氮键成键反应的研究也取得了长足进步,尤其是对氮链延长反应的研究。
氨基的氧化偶联是制备偶氮的常用方法,N—NH2的偶联反应可以用来构建四氮烯结构,是延长氮链的常用手段。1985年,Neunhoeffer等[11-12]将1‐氨基吡唑用NiO2及AcOH处理,得到偶联产物1,1′ ‐偶氮吡唑,该化合物具有碳取代的N6链结构。2007年,Yu‐Chuan Li等[13]用二氯异氰尿酸钠(SDCI)和乙酸,合成了1,1′ ‐偶氮‐1,2,3‐三唑,成功构建了碳取代的N8链结构。2011年,Thomas M等[14]用类似方法合成了含有N10链的偶氮四唑化合物1,1′‐偶氮‐四唑。
重氮盐正离子可以和芳胺上的氮原子发生偶联反应,生成重氮‐氨基结构,也是延长氮链的一种方法。Bubnov P F[15-16]报道5‐氨基四唑形成的重氮盐能继续和5‐氨基四唑反应,生成5,5′‐重氮胺四唑。杨红伟等[17]将1,5‐二氨基四唑用盐酸和亚硝酸钠处理,构建了碳取代的N11链结构。
氮烯是一类具有— N = N —、— N = N — N或—N=N—N=N—结构的分子,和烯烃在碳骨架构建中的广泛应用相比,目前氮烯在氮骨架合成中的研究很少。N. Wiberg等[18]曾报道通过热解1,2‐二(三甲基硅基)二氮烯能合成四(三甲基硅基)四氮烯,由于硅基极易通过N—Si键的酸解而脱去,因此,该类反应在氮骨架构建中有潜在的应用,有必要对其进行进一步研究。
为此,本研究通过15N同位素标记实验分析了1,2‐二(三甲基硅基)二氮烯的形成历程,研究了不同温度下1,2‐二(三甲基硅基)二氮烯的热解反应,并通过量子化学计算分析了1,2‐二(三甲基硅基)二氮烯在高温下生成1,1,4,4‐四(三甲基硅基)四氮烯的反应机理,为氮烯的该类反应提供借鉴。
2 实验部分 2.1 试剂与仪器试剂:叠氮化钠、无水硫酸钠、无水氯化钙、金属钠,分析纯,成都市科龙化工试剂厂;乙醇、正戊烷、无水乙醚,分析纯,天津化学试剂三厂;对甲苯磺酰氯、无水硫酸钠、15N标记叠氮化钠、正丁基锂,分析纯,美国百灵威科技科技有限公司;无水乙醚经无水氯化钙干燥过夜,加钠丝回流,惰性气体保护下蒸出待用;1,1,2‐三(三甲基硅基)肼,按[19]文献自制。
仪器:JA 5003B型分析天平,上海精密科学仪器有限公司;AV500型(500 MHz)超导核磁共振仪,瑞士Bruker公司;QP‐2010 Plus型气质联用仪,日本岛津公司;5700 FT‐IR型傅里叶变换红外光谱仪,美国热电尼高力公司;Cary 60型紫外‐可见吸收光谱仪,美国Agilent公司;JH350显微热台熔点仪,上海佳航仪器仪表有限公司。
2.2 计算方法所有计算均采用Gaussian 09程序[20]完成。在B3LYP / 6‐31G(d)水平上得到反应物、产物、中间体和过渡态的几何构型,通过在B3LYP/6‐31G(d)水平上进行的振动频率分析表明,中间体的振动频率都是正值,过渡态只有一个虚频。
2.3 合成路线以三(三甲基硅基)肼为起始原料,合成三(三甲基硅基)肼锂,然后通过和对甲苯磺酰叠氮反应,合成了高活性的1,2‐二(三甲基硅基)二氮烯,进一步利用溶剂热反应,合成了1,1,4,4‐四(三甲基硅基)四氮烯。合成路线如Scheme 1所示。

|
Scheme1 Synthetic route for 1, 1, 4, 4-tetrakis(trimethylsi- lyl) tetrazene |
氩气保护下,将1,1,2‐三(三甲基硅基)肼(24.8 g,100 mmol)加入250 mL圆底烧瓶中,冰浴冷却,搅拌下滴加1.3 mol·L-1的正丁基锂戊烷溶液(85 mL,110 mmol),反应1 h,然后冷却至-40 ℃,在氩气手套箱中快速趁冷过滤,得白色固体23.4 g,收率92%,于无水无氧条件下保存。1H NMR(500 MHz,C2D5OC2D5)δ:0.07(s,27H,CH3)。
2.4.2 对甲苯磺酰叠氮的合成将叠氮化钠(15.6 g,240 mmol)溶于50 mL去离子水和50 mL乙醇的混合溶剂中,室温搅拌,缓慢滴加对甲苯磺酰氯(38.1 g,200 mmol)的饱和乙醇溶液,加毕,继续反应1 h,减压蒸馏除去乙醇。将残余物用去离子水洗涤3次,收集有机相,用无水硫酸钠干燥过夜,抽滤得无色液体36.6 g,收率93%。FT ‐IR(KBr,ν/cm-1):2121(N≡N),1595(C = C),1366(S= O),1161(S= O),1084(C—H),812(C—H),743(C—H),656,587,537;1H NMR(500 MHz,CDCl3)δ:7.75(d,2H,CH),7.35(d,2H,CH),2.34(s,3H,CH3);Anal. calcd for C7H7SO2N3:C 42.64,H 3.55,N 21.32;Found:C 42.86,H 3.61,N 21.26。
15N标记的对甲苯磺酰叠氮以15N标记的叠氮化钠为原料,合成方法同上。
2.4.3 1,2-二(三甲基硅基)二氮烯的合成氩气保护下,将1,1,2 ‐三(三甲基硅基)肼锂(25.4 g,100 mmol)和200 mL无水乙醚加入500 mL三口烧瓶中,搅拌,冷却至-78 ℃,缓慢滴加1.5 mol·L-1的对甲苯磺酰叠氮无水乙醚溶液(67 mL,100.5 mmol),滴加完毕后继续反应2 h,得深绿色溶液。升温至-50 ℃,接液氮冷阱,减压,除去乙醚,然后快速升温至-5 ℃,用另一干冰冷阱减压冷凝,得淡蓝色固体,充氩气,缓慢升温得淡蓝色液体10.1 g,收率58%,于- 40 ℃下且无水无氧环境保存。1H NMR(500 MHz,CD3Cl)δ:0.16(s,18H,CH3);UV‐Vis(λmax,ether):786 nm。
2.4.4 1,2-二(三甲基硅基)二氮烯合成过程中气体产物分析向上述1,2‐二(三甲基硅基)二氮烯合成装置接气体收集袋,用氩气抽换气三次。氩气保护下,将1,1,2‐三(三甲基硅基)肼锂(25.4 g,100 mmol)和200 mL无水乙醚加入500 mL三口烧瓶中,搅拌,冷却至- 78 ℃,打开气体收集袋,缓慢滴加1.5 mol · L-1的15N标记对甲苯磺酰叠氮的无水乙醚溶液(67 mL,100.5 mmol),滴加完毕后继续反应2 h。将气体收集袋密封,用GC‐MS进行表征。
2.4.5 1,1,4,4-四(三甲基硅基)四氮烯的合成氩气保护下,将1,2 ‐二(三甲基硅基)二氮烯(10.1 g,58 mmol)和60 mL无水无氧苯快速加入干燥的100 mL Pyrex管中,密封,放入150 ℃的油浴中反应1 h,用微型精馏装置减压精馏,收集72~75 ℃馏分,将所得混合物用无水正戊烷在-78 ℃重结晶,获得白色粘稠固体0.94 g,收率9.4%,熔点37~38 ℃。FT ‐ IR(KBr,ν/cm-1):2960(C—H),2919(C—H),2854(C—H),1253(N = N),1075,929(Si—N),840(N—N),753(C—H),438;1H NMR(500 MHz,C6D6)δ:0.06(s,36H,CH3);Anal. calcd for C12H36N4Si4:C 41.35,H 10.33,N 16.08;Found:C 41.77,H 11.02,N 15.95。
3 结果与讨论 3.1 硅基取代二氮烯的合成氨基的氧化偶联是合成偶氮键的常用方法,但硅胺类化合物很少有类似反应的报道。本研究以三苯基硅胺、三异丙基硅胺为底物,通过氧化汞、对苯醌、二氯异氰尿酸钠等进行氧化,都未得到上述硅胺化合物的氧化偶联产物。
以三(三甲基硅基)肼锂为底物,按照文献[21],通过和对甲苯磺酰叠氮反应,合成了1,2‐二(三甲基硅基)二氮烯(Scheme 1)。对甲苯磺酰叠氮较温和的氧化剂,它易与三(三甲基硅基)肼锂上的锂氮反应,氧化N—N单键生成N=N双键,并使反应停留在N=N双键的阶段,而不直接氧化成N≡N叁键(即氮气)。该反应是一个强放热、快速释放气体的过程,检测反应历程比较困难。Wiberg N等[22]认为,该反应可能经历了一个长链N5的过程,如Scheme 2所示,长链N5先分解放出氮气,然后进一步消去三甲基硅基‐对甲苯磺酰氨基锂形成氮氮双键,生成1,2‐二(三甲基硅基)二氮烯。

|
Scheme2 Reaction path for preparation of bis(trimethylsilyl)diimine[22] |
为分析该历程,以15N标记的叠氮化钠(叠氮基的一个末端N被标记)为原料,合成了15N标记的对甲苯磺酰叠氮,然后和三(三甲基硅基)肼锂反应,收集反应过程中放出的气体,并通过GC‐MS进行表征。15N标记的对甲苯磺酰叠氮(NαNβNγ‐Tos)中15N标记位点可能为Nα或Nγ,且含量相同,两种情况下分别形成长链N5结构,如Scheme 2所示,分别分解放出等量的N≡N(m/z 28)和15N≡N(m/z 29)。实验中获得的典型质谱图如图 1所示,除了大量的氩气外(m/z 40),发现了分子量为28和29的氮气,且二者的丰度比基本为1:1,与文献[22]所述机理推测的结果相一致。

|
图 1 1,2-二(三甲基硅基)二氮烯合成反应气体产物质谱图 Fig.1 MS spectrum of gas products in preparation of 1, 2-bis (trimethylsilyl)diimine |
1,2‐二(三甲基硅基)二氮烯对空气、水极其敏感,而且自身稳定性差[22],因此整个实验过程必须严格控制无水无氧,且始终保持在较低温度下。产物的这些特性决定了难以使用常规的分离提纯方法,利用二氮烯和系列副产物沸点的差异,在高真空条件下对二氮烯进行低温冷凝,实现了其纯化。
3.2 硅基取代四氮烯的合成1,2‐二(三甲基硅基)二氮烯在0 ℃就会分解放出氮气,将其溶液在氩气保护下室温放置,待彻底分解后进行GC‐MS表征,发现其中含有四(三甲基硅基)肼、三(三甲基硅基)肼和二(三甲基硅基)胺等产物,如Scheme 3所示,在此条件下基本难以发现二聚产物。

|
Scheme3 Pyrolysis products of bis(trimethylsilyl)diimine at different temperatures |
将1,2‐二(三甲基硅基)二氮烯在氩气保护下密封于Pyrex管中,迅速置于150 ℃的油浴中反应,反应产物中除了大量上述化合物外,还有少量1,1,4,4‐四(三甲基硅基)四氮烯生成,收率约9.4%。虽然1,1,4,4‐四(三甲基硅基)四氮烯收率仍然较低,但上述实验证明高温条件有利于1,2‐二(三甲基硅基)二氮烯转化为1,1,4,4‐四(三甲基硅基)四氮烯。
3.3 硅基取代四氮烯反应机理的理论研究由B3LYP/6‐31G(d)方法优化的1,2‐二(三甲基硅基)二氮烯和四(三甲基硅)四氮烯的结构如图 2所示。1,2‐二(三甲基硅基)二氮烯分子中的Si—N=N—Si四个原子处于同一平面,N=N双键键长为1.260 Å,N—Si单键键长为1.827 Å,Si—C单键键长为1.888 Å,Si—N=N键角为116.1°。1,1,4,4‐四(三甲基硅)四氮烯的二面角∠N(1)N(2)N(3)N(4)为-175.0°,四个氮原子为处于近平面状态的链式结构;二面角∠Si(1)N(1)N(2)N(3)为18.1°,二面角∠N(2)N(3)N(4)Si(4)为-163.7°,因此Si(1)N(1)N(2)N(3)N(4)Si(4)为处于非平面状态的链式结构;N(2)=N(3)双键键长为1.254 Å,N(1)—N(2)和N(3)—N(4)两个单键键长分别为1.398 Å和1.397 Å。

|
图 2 1,2-二(三甲基硅基)二氮烯和1,1,4,4-四(三甲基硅基)四氮烯的优化结构 Fig.2 Optimized structures of 1, 2-bis (trimethylsilyl) diimine (BSD)and 1, 1, 4, 4-tetrakis (trimethylsilyl) tetrazene(TST) |
W. R. Mcbride等[23]报道1,1‐二烷基二氮烯能二聚形成四烷基四氮烯。1,2‐二(三甲基硅基)二氮烯(BSD)热解为1,1,4,4‐四(三甲基硅)四氮烯(TST)的反应也可能经历了1,1 ‐二硅基二氮烯的过程,如Scheme 4所示:1,2‐二(三甲基硅基)二氮烯分子中一个三甲基硅基向另一端N原子靠近,经过渡态TS1形成了1,1‐二(三甲基硅基)二氮烯中间体IM1;两个中间体IM1相互作用,成键,经过渡态TS2形成了1,1,4,4‐四(三甲基硅)四氮烯。

|
Scheme4 Reaction path for preparation of 1, 1, 4, 4-tetrakis (trimethylsilyl)tetrazene |
对上述过程中各中间体、过渡态的能量进行了计算,如Scheme 5所示,结果表明:BSD需要克服103.0 kJ·mol-1的活化能才能形成过渡态TS1,BSD转化为中间体IM1需要从外界吸热78.7 kJ·mol-1;中间体IM1进一步越过114.3 kJ·mol-1的活化能生成TST,同时向外释放169.9 kJ·mol-1的内能。总体来讲,1,2‐二(三甲基硅基)二氮烯反应生成1,1,4,4‐四(三甲基硅)四氮烯需要高达193.0 kJ·mol-1的活化能,而反应完成后释放91.2 kJ·mol-1的热量。因此,该反应整体上为放热过程,但需要高温活化以便越过高能过渡态,这和3.2节中高温条件有利于该反应发生的实验结果一致。

|
Scheme5 Potential energy profiles at the B3LYP/6-31G(d) level (unit:kJ·mol-1) |
(1)三(三甲基硅基)肼锂与对甲苯磺酰叠氮反应,合成了1,2‐二(三甲基硅基)二氮烯;研究了1,2‐二(三甲基硅基)二氮烯在不同温度下的热解反应,发现高温条件有利于其生成1,1,4,4‐四(三甲基硅基)四氮烯,溶剂热反应中1,1,4,4‐四(三甲基硅基)四氮烯的收率约为9.4%。
(2)用量子化学计算方法研究了该反应的机理,发现1,2‐二(三甲基硅基)二氮烯先异构化为1,1‐二(三甲基硅基)二氮烯中间体,两个中间体相互作用形成1,1,4,4‐四(三甲基硅基)四氮烯,两个过程分别需要103.0 kJ·mol-1和114.3 kJ·mol-1的活化能,该结果与实验现象一致。
| [1] |
Lauderdale W J, Stanton J F, Bartlett R J. Stability and energetics of metastable molecules: Tetraazatetrahedrane (N4), hexaazabenzene (N6), and octaazacubane (N8)[J].
The Journal of Physical Chemistry, 1992, 96(3): 1173-1178. DOI:10.1021/j100182a029 |
| [2] |
Östmark H. High energy density materials (HEDM):Over‐ view, theory and synthetic efforts at FOI[J].
New Trends in Research of Energetic Materials Czech Republic, 2006: 231-250. |
| [3] |
Samartzis P C, Wodtke A M. All‐nitrogen chemistry:How far are we from N60?[J].
International Reviews in Physical Chemistry, 2006, 25(4): 527-552. DOI:10.1080/01442350600879319 |
| [4] |
Glukhovtsev M N, Jiao H, Schleyer P V R. Besides N2, what is the most stable molecule composed only of nitrogen atoms?[J].
Inorganic Chemistry, 1996, 35(24): 7124-7133. DOI:10.1021/ic9606237 |
| [5] |
Christe K O, Wilson W W, Sheehy J A, et al. N5+ :A novel homolepticpolynitrogen ion as a high energy density material[J].
Angewandte Chemie International Edition, 1999, 38(13- 14): 2004-2009. |
| [6] |
Vij A, Wilson W W, Vij V, et al. Polynitrogenchemistry.Syn thesis, characterization, and crystal structure of surprisingly stable fluoroantimonatesalts of N5+[J].
Journal of the American Chemical Society, 2001, 123(6): 6308-6313. |
| [7] |
Vij A, Pavlovich J G, Wilson W W, et al. Experimental detection of the pentaazacyclopentadienide(pentazolate)anion, cyclo‐N5-[J].
Angewandte Chemie International Edition, 2002, 41(16): 3051-3054. DOI:10.1002/1521-3773(20020816)41:16<3051::AID-ANIE3051>3.0.CO;2-T |
| [8] |
Bazanov B, Geiger U, Carmieli R, et al. Detection of cyclo ‐ N5- in THF solution[J].
Angewandte Chemie International Edition, 2016, 55(42): 13233-13235. DOI:10.1002/anie.201605400 |
| [9] |
Zhang C, Sun C G, Hu B C, et al. Synthesis and characterization of the pentazolate anion cyclo ‐ N5‐ in (N5) 6(H3O) 3 (NH4) 4Cl[J].
Science, 2017, 355(6323): 374-376. DOI:10.1126/science.aah3840 |
| [10] |
Xu Y G, Wang Q, Shen C, et al. A series of energetic metal pentazolatehydrates[J].
Nature, 2017, 549(7670): 78-81. DOI:10.1038/nature23662 |
| [11] |
Neunhoeffer H, Clausen M, Vötter H‐D, et al. 1, 2, 3‐Triazine, Ⅲ Synthese von N ‐aminopyrazolen und derenoxidation zu 1, 2, 3 ‐ triazinen. Molekülstruktur des 1, 2, 3 ‐ triazins[J].
European Journal of Organic Chemistry, 1985, 1985(9): 1732-1751. |
| [12] |
Ohsawa A, Arai H. Oxidation of 1 ‐ aminopyrazoles and synthesis of 1, 2, 3 ‐ triazines[J].
The Journal of Organic Chemis- try, 1985, 50(26): 5520-5523. DOI:10.1021/jo00350a017 |
| [13] |
Li Y C, Qi C, Li S H, et al. 1, 1'‐Azobis‐ 1, 2, 3 ‐triazole:A high ‐ nitrogen compound with stable N8 structure and photo‐ chromism[J].
Journal of the American Chemical Society, 2010, 132(35): 12172-12173. DOI:10.1021/ja103525v |
| [14] |
Klapötke T M, Piercey D G. 1, 1'‐Azobis(tetrazole):A highly energetic nitrogen ‐ rich compound with a N10 chain[J].
Inorganic Chemistry, 2011, 50(7): 2732-2734. DOI:10.1021/ic200071q |
| [15] |
Bubnov P F.
Primary Explosives and Initiation Devices[M]. Part 1. Moscow: Drofa Press, 1940: 311-314.
|
| [16] |
Khmelnitskij L I.
Handbook of Explosive Materials[M]. Part 2. Moscow: Drofa Press, 1961: 89-90.
|
| [17] |
Tang Y X, Yang H W, Wu B, et al. Synthesis and characterization of a stable catenated N11 energetic salt[J].
Angewandte Chemie International Edition, 2013, 52(18): 4875-4877. DOI:10.1002/anie.v52.18 |
| [18] |
Wiberg N, UhlenbrockW. 1, 1, 4, 4‐Tetrakis (trimethylsilyl) tetrazene[J].
Angewandte Chemie International Edition, 1970, 9(1): 70-71. DOI:10.1002/(ISSN)1521-3773 |
| [19] |
丁可伟, 李陶琦, 肖啸, 等. 三甲基硅基肼的合成研究[J].
含能材料, 2017, 25(6): 498-502. DING Ke‐wei, LI Tao‐qi, XIAO Xiao, et al. Synthesis studies of trimethylsilylhydrazine[J]. Chinese Journal of energetic materials(Hanneng Cailiao), 2017, 25(6): 498-502. DOI:10.11943/j.issn.1006-9941.2017.06.009 |
| [20] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09 [CP], Gaussian, Inc., Wallingford, CT, 2009
|
| [21] |
Wiberg N, Joo W ‐Ch, Uhlenbrock W. Bis(trimethylsilyl)diimine and tetrakis(trimethylsilyl)hydrazine[J].
Angewandte Chemie International Edition, 1968, 7(8): 640-640. |
| [22] |
Wiberg N. Bis(trimethylsilyl)diimine:preparation, structure, and reactivity[J].
Angewandte Chemie International Edition, 1971, 10(6): 374-387. DOI:10.1002/(ISSN)1521-3773 |
| [23] |
Mcbride W R, Bens E M. Alkylhydrazines. Ⅲ. Dimerization of certain substituted 1, 1 ‐ dialkyldiazenes to tetraalkyltetrazenes[J].
Journal of the American Chemical Society, 1959, 81(21): 5546-5550. DOI:10.1021/ja01530a008 |
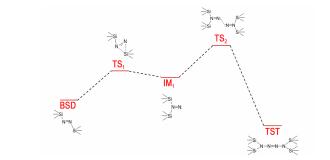
Tetra(trimethylsilyl)tetrazene was synthesized via dimerization reaction of 1,2‐bis(trimethylsilyl)diimine. The DFT calculation shows that this reaction involves two steps:1,2‐bis(trimethylsilyl)diimine is firstly isomerized to 1,1‐bis(trimethylsilyl)diimine intermediate and then two 1,1‐bis(trimethylsilyl)diimine intermediates coupled to form tetra(trimethylsilyl)tetrazene.