2. 航天科工集团江河化工厂,湖北 远安 444200
2. State Jianghe Chemical Plant of CASIC, Yuanan 444200, China
键合剂是复合推进剂中用于改善粘合剂和固体颗粒之间界面作用的化合物。键合剂的种类很多,主要有醇胺、钛酸酯、有机硅烷、硼酸酯、氮丙啶等小分子键合剂[1-2];海因/三嗪等复配键合剂[3];海因基高分子[4]、含不同取代的三聚氰酰聚醚醇、三聚异氰脲聚醚、中性聚合物[5-6]等大分子键合剂。
不同的固体填料与粘合剂体系,使用的键合剂分子结构也不同[7]。比如,N, N'-双(2-叠氮乙基)草酰胺作为HMX的键合剂[8]。硼酸酯类键合剂主要用于改善硝胺复合推进剂的高低温力学性能[9]。硼酸酯键合剂中B原子的sp2杂化轨道能与RDX中电子供体(如N原子)形成稳定的配合物,有利于硼酸酯结合在硝胺颗粒上。此外,该类键合剂分子结构中还含有羟基,使其能与固化剂反应进入粘合剂网络,在硝胺颗粒表面形成高模量层。
AP/RDX/Al/HTPB推进剂中硝胺开发设计的硼酸酯类键合剂已被证明具有良好的抗硝胺脱湿能力,可有效改善了该类推进剂的力学性能[9-11]。由于对其键合作用机理的认识还不够,需要从分子层次上来研究键合剂的结构、分子轨道及其与RDX的界面相互作用等问题。为此,本研究通过理论计算,探讨了5种新研制的硼酸酯键合剂分子结构及其与RDX键合作用机理。从硼酸酯的分子构象入手,计算了分子轨道;再通过分子动力学模拟计算硼酸酯键合剂与RDX两种晶面的结合能,分析了键合作用机理。
2 硼酸酯键合剂的结构特征一些传统键合剂如MAPO等对AP/HTPB复合推进剂体系有效,但对RDX/HTPB界面却无效。在四组元推进剂中键合剂不仅要与硝胺、AP等键合,而且还需要与固化剂反应成为粘合剂的一部分。为了提高键合剂的综合应用性,在AP/RDX/A国/HTPB推进剂中考虑应用硼酸酯类键合剂[9-10]。这种键合剂分子中的羟基能与异腈酸酯反应,形成的强极性—CONH官能团又可以与RDX中的NO2基团形成氢键;此外,在理论上硼酸酯中B原子的sp2杂化轨道能与RDX中电子供体硝胺基团形成稳定的络合物,有利于硼酸酯包覆在硝胺颗粒上。本课题组设计并合成的5种硼酸酯键合剂分子均具有双硼酸酯结构,旨在加强B原子与RDX相互作用的研究。5种硼酸酯键合剂结构如图 1所示,其特征取代基见表 1。

|
图 1 硼酸酯键合剂的分子结构图(图中R1、R2和R3均为烷基) Fig.1 Molecular structure of borate ester bonding agent (R1, R2 and R3 are alkyls) |
| 表 1 硼酸酯键合剂的特征取代基 Tab.1 Characteristic groups of borate ester bonding agent |
RDX的实验数据[12]表明,RDX的主要晶面为(210)和(200)。在分子模拟过程中,选取这两个有代表性、且所占晶面比例较大的晶面进行研究。RDX晶胞的初始结构由实验数据获得,在此基础上切割构建RDX(210)和RDX(200)晶面,然后再构建RDX切面超晶胞(图 2)。RDX(210)和RDX(200)晶面的切割厚度均为2.0 nm。硼酸酯键合剂与RDX晶胞的界面模型构建方法为:首先建立BEBA_1~5无定型模型截面,然后将RDX晶面和BEBA分子构建为上下两层的表面粘附模型(RDX(210)/BEBA和RDX(200)/BEBA相互作用模型),经过分子动力学(MD)过程驰豫得到界面模型。建模使用了Material Studio软件的Visualizer模块。

|
图 2 RDX(210)和RDX(200)晶面 Fig.2 RDX supercells with (210) and (200) crystal surface |
对5种键合剂的构象搜索采用的是退火分子动力学方法。在300~3000 K温度范围内进行100次退火循环(共105步),NVT系综和Nosé热浴。对退火分子动力学每次循环所获得的结构都进行能量优化,选取能量最低的作为量化计算的初始结构和MD模拟的分子结构。使用杂化电子密度泛函B3LYP(Becke三参数交换函数与Lee-Yang-Parr相关函数)结合3-21G(d)基组对所获得的初始结构进行量化计算。计算在Gaussian 03程序上完成[13]。
在模拟相互作用能的过程中,由于键合剂一般只同RDX表面的分子发生相互作用,所以在模拟相互作用能的过程中,固定RDX下层分子坐标仅保留上面一层分子进行分子动力学驰豫过程。分子动力学过程采用Dreiding力场,NVT系综、Nosé热力浴,时间步长1 fs,在298K下完成。对体系分配Qeq电荷[14],库仑非键相互作用使用Ewald加和法,范德华相互作用采用原子加和法,截断半径为1.25 nm。对分子动力学平衡后模型结构进行单点能计算,分析界面之间的相互作用能大小,在单点能计算过程中解除固定的原子坐标,具体的计算公式为:
| $ \Delta {{E}_{\rm{inter}}}={{E}_{\rm{total}}}-\left( {{E}_{\rm{RDX}}}+{{E}_{\rm{BEBA}}} \right) $ | (1) |
式中,ΔEinter为相互作用能;Etotal为RDX/BEBA体系的能量;ERDX为RDX体系的能量;EBEBA为BEBA的能量;定义BEBA/RDX之间的结合能为Ebind=-Einter。
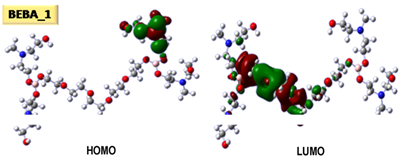
4 模拟计算结果 4.1 硼酸酯键合剂分子结构及分子轨道运用退火分子动力学方法获得了5种硼酸酯键合剂分子的稳定构象如图 3所示。

|
图 3 硼酸酯、水和硼酸甲基最稳定构象以及它们的HOMO和LUMO图 Fig.3 Most stable conformation of borate esters, water and trimethyl borate and their HOMO and LUMO images |
由图 3可以看出,BEBA_2和BEBA_5分子侧链上含有较大基团,会造成内旋转困难,使分子柔性变差;而BEBA_1和BEBA_3柔性较好,预测有利于消除推进剂拉伸过程中在填料与基体界面的应力集中。BEBA_1和BEBA_2轨道能隙较大(分别为7.08和6.25 eV),说明它们具有相对较好的电子稳定性。5种键合剂BEBA_1、BEBA_2、BEBA_3、BEBA_4和BEBA_5的偶极矩依次为5.37、6.65、11.89、11.97和7.88 Debye。偶极矩较大说明其极性大。较大的偶极矩会产生较大的极化率,使得分子诱导力增强,有利于键合作用。因此,预计BEBA_4、BEBA_3和BEBA_5与RDX之间有较强的键合作用。
硼酸酯键合剂理论上可以通过硼原子的2p空轨道与推进剂填料分子的负电性基团产生络合相互作用。对比BEBA分子和硼酸甲酯的HOMO和LUMO轨道图可以看出,这种作用对小分子硼酸酯更容易实现,相对于本文研究对象——双硼酸酯键合剂而言,小分子硼酸酯的最低未占据分子轨道(LUMO)更容易处于硼原子位置处。但是小分子硼酸酯会出现类似于烷基钛酸酯的水解问题。设计合成的大分子硼酸酯键合剂通过提高硼原子所处空轨道的能级(如图 3中BEBA_1的LUMO+2轨道),不仅降低了H2O的最高占据轨道(HOMO)进攻B的几率,同时也一定程度上降低了BEBA与RDX形成复合相互作用的几率。但双硼酸酯键合剂结构有利于改善BEBA与RDX之间的相互作用。
图 3表明5种硼酸酯键合剂中硼酸酯基团所处最低空轨道的能级均比较高,不利于硼酸酯中硼的空轨道与H2O产生作用。此外,预计与硝胺分子相互作用也主要以范德华、静电和氢键相互作用为主。因此,可以通过分子模拟来研究硼酸酯键合剂与RDX之间的相互作用机理。
4.2 硼酸酯键合剂与RDX不同晶面的结合能采用分子动力学模拟方法,研究了5种键合剂分别与RDX(210)、RDX(200)晶面的相互作用能。图 4分别为5种硼酸酯键合剂与RDX (210)晶面的结合模型。键合剂与RDX (200)晶面的结合模型类似,本文不再赘述。从图 4可以看出,经过分子动力学驰豫平衡之后,硼酸酯能够很好地粘附在RDX表面上。

|
图 4 RDX (210)面与五种键合剂的结合模型 Fig.4 Bonding models for RDX (210) surface and five bonding agents |
表 2和表 3分别给出了RDX (210)和RDX (200)晶面与硼酸酯结合的相互作用能。结果表明,5种硼酸酯键合剂对RDX都有键合作用;相互作用能主要来自范德华力和静电相互作用;整个体系都存在氢键相互作用但贡献较小。相比小分子硼化物(如BH3、BF3等)与硝胺中NO2上的O形成的B…O强相互作用,本文键合剂的大分子柔性结构使空间位阻增加,从而增大了B…O距离,一定程度上降低了B…O相互作用。
| 表 2 硼酸酯键合剂在RDX(210)面上的结合能 Tab.2 Binding energy for five BEBAs on the surface of RDX(210) surface |
| 表 3 硼酸酯键合剂在RDX(200)面上结合能 Tab.3 Binding energy for five BEBAs on the surface of RDX(200) surface |
硼酸酯键合剂BEBA_1与RDX(210)晶面结合最好,BEBA_5与RDX(210)晶面结合能力一般。所有RDX(210)/BEBA结合模型中静电相互能都大于范德华相互作用能。RDX(210)/BEBA结合的总键合能力顺序为RDX(210)/BEBA_1>RDX(210)/BEBA_4≈RDX(210)/BEBA_2>RDX(210)/BEBA_3>RDX(210)/BEBA_5。
BEBA_1和BEBA_2连接的R4是供电子烷烃基,使硼酸酯键合剂分子中胺氮的负电荷增加,在诱导作用下增大了RDX分子上NO2上O和N之间的电荷差异,提高了彼此之间的相互作用能对结合能的贡献。虽然BEBA_2中正丁基给电子能力比BEBA_1中甲基强,但空间位阻效应降低了其相互作用。硼酸酯BEBA_3和BEBA_5中R4分别为吸电子的乙酰基和甲酰丙酮基,使胺氮的负电性减小,造成O…N相互极化作用减小,降低了NO2与胺氮之间相互作用能对总结合能的贡献;但有利于B原子空轨道与RDX负电性基团形成配位键的几率。BEBA_4虽然与RDX分子中N…O相互作用降低,但RDX分子中NO2上O的电负性大于BEBA_4中CN基上的N,腈基与NO2基团发生诱导效应使相互作用增强。所以RDX(210)/BEBA_4也有较大的结合能。
对比表 2和表 3结果表明,BEBA与RDX(200)晶面的结合能大于其与RDX(210)晶面的结合能,说明硼酸酯键合剂在RDX晶面上的键合作用具有选择性;RDX(200)/BEBA相互作用中氢键的贡献同RDX(210)/BEBA相互作用中的基本相同,且都为不是主要贡献项。氢键主要都是RDX晶面上NO2与硼酸酯上OH基团形成的ONO…HO键;相互作用能也都主要来自非键相互作用中的静电相互作用和范德华相互作用。硼酸酯键合剂与RDX(200)晶面结合能顺序为RDX(200)/BEBA_5>RDX(200)/BEBA_2>RDX(200)/BEBA_1>RDX(200)/BEBA_3>RDX(200)/BEBA_4。
即BEBA_5和BEBA_2键合能力较好。BEBA_5在RDX(210)和RDX(200)晶面上的相对结合能差异很大。RDX(200)/BEBA_5体系中结合能最大,推测是由于RDX(200)晶面最表层氧原子含量(6.5个/nm2)比RDX(210)晶面最表层氧原子含量(10个/nm2)少,有利于甲酰丙酮基上有两个羰基对RDX上的NO2基团产生诱导效应,产生强的结合作用,抵消并补偿了因BEBA_5中胺氮负电荷减少而降低的结合能。此外,甲酰丙酮基较大的空间位阻也使得RDX(200)/BEBA_5之间的相互作用更多地来自羰基与NO2之间的诱导相互作用。由此可见,在RDX(210)/BEBA_5体系中,RDX(210)晶面最表层过多氧原子不利BEBA_5中的羰基与其作用,同时由于BEBA_5中胺氮负电荷减少,碱性变弱,造成了结合能的降低。所以,在应用硼酸酯键合剂键合硝胺过程中,不仅要考虑键合剂的取代基、空间位阻的影响,还要考虑硝胺晶面结构特征的影响。
5 结论通过对5种硼酸酯键合剂分子结构、分子轨道的理论计算,以及运用分子动力学方法对其与RDX(210)和RDX(200)晶面相互作用的研究,得到以下结论:
(1) 5种BEBA分子硼原子所处空轨道的能级较高,不利于硼酸酯中硼的空轨道与H2O和与硝胺分子产生很强的化学作用。双硼酸酯键合剂结构有改善硼酸酯与RDX的键合作用。键合剂与RDX的键合作用以范德华和静电相互作用为主。
(2) BEBA与RDX(200)晶面的结合能大于其与RDX(210)晶面的结合能,BEBA分子与RDX晶面相互作用具有选择性。总体上,BEBA_1、BEBA_2与RDX晶面的键合作用较强。
(3) BEBA_5在RDX(210)和RDX(200)晶面上的结合能力相反。在RDX(200)/BEBA_5中结合最好,推测是因RDX(200)晶面最表层氧原子含量比RDX(210)晶面最表层氧原子含量少,有利于甲酰丙酮基上有两个羰基对RDX上的NO2基团产生诱导效应,产生强的结合作用,抵消并补偿了因BEBA_5中胺氮负电荷减少而降低的结合能。而RDX(210)/BEBA5体系中,RDX(210)晶面最表层过多氧原子不利于BEBA_5与其作用。
| [1] |
王一颖, 邱刚. 键合剂1, 1'-(1, 3-苯二羰基)双-2-甲基氮丙啶的热行为[J].
推进技术, 2001, 22(3): 258-260. WANG Yi-ying, QIU Gang. Thermal behavior of bonding agent 1, 1'-(1, 3-phenylenedlcarbonyl) bis-2-methyl-aziridine[J]. Journal of Propulsion Technology, 2001, 22(3): 258-260. |
| [2] |
Gercel B O, Vner D O, Pekel, et al. Improved adhesive properties and bonding performance of HTPB-based polyurethane elastomer by using aziridine-type bond promoter[J].
J Appl Polym Sci, 2001, 80: 806-814. DOI:10.1002/(ISSN)1097-4628 |
| [3] |
李江存, 焦清介, 任慧, 等. 海因/三嗪类复合键合剂包覆黑索今的研究[J].
含能材料, 2008, 16(1): 56-59. LI Jiang-cun, JIAO Qing-jie, REN Hui, et al. RDX coated with hyantoin/triazines composite bonding agent[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 2008, 16(1): 56-59. |
| [4] |
王延铭, 贾秀兰, 孙翔宇, 等. 几种5, 5-二甲基海因高分子键合剂的合成及其性能研究[J].
内蒙古科技大学学报, 2007, 26(4): 362-364. WANG Yan-ming, JIA Xiu-lan, SUN Xiang-yu, et al. The synthesis of seve ral macromolecule bonding agents containing 5, 5-dimethyl hydantoin and the study on their properties[J]. Journal of Inner Mongolia University of Science and Technology, 2007, 26(4): 362-364. |
| [5] |
王北海. 中性聚合物键合剂的分子设计和合成[J].
推进技术, 1995, 16(4): 71-76. WANG Bei-hai. Molecular design and synthesis of neutral polymeric bonding[J]. Journal of Propulsion Technology, 1995, 16(4): 71-76. |
| [6] |
雷贝, 邓剑如, 陈浪, 等. NPBA的水分散聚合研究[J].
火炸药学报, 2008, 31(1): 60-63. LEI Bei, DENG Jian-ru, CHEN Lang, et al. Study on water dispersion polymerization of NPBA[J]. Chinese Journal of Explosives & Propellants, 2008, 31(1): 60-63. |
| [7] |
杜磊, 肖金武, 尹瑞康. 高燃速HTPB/IPDI推进剂低温力学性能(Ⅱ)界面助剂的设计与应用[J].
推进技术, 2002, 23(3): 245-248. DU Lei, XIAO Jin-wu, YIN Rui-kang. Low temperature mechanical properties of high-burning rate HTPB/IPDI propellants[J]. Journal of Propulsion Technology, 2002, 23(3): 245-248. |
| [8] |
董树安, 欧育湘. N, N'-双(2-叠氮乙基)草酰胺的合成及性能[J].
含能材料, 1997, 5(4): 157-161. DONG Shu-an, OU Yu-xiang. Synthesis and properties of N, N'-bis(2-azidoethyl) oxalic amide[J]. Chinese Journal of Energetic Materials (Hanneng Cailiao), 1997, 5(4): 157-161. |
| [9] |
彭网大, 王春华, 张仁. 硼化物键合剂在硝胺推进剂中的应用研究[J].
固体火箭技术, 1990, 13(4): 43-49. PENG Wang-da, WANG Chun-hua, ZHANG Ren. Study on application of the boron compound bonding agents in the Nitramine propellants[J]. Journal of Solid Rocket Technology, 1990, 13(4): 43-49. |
| [10] |
崔瑞禧, 张炜, 邓剑如. 硼酸酯键合剂在丁羟四组元推进剂中作用机理研究[J].
推进技术, 2010, 31(4): 448-451. CUI Rui-xi, ZHANG Wei, DENG Jian-ru. Mechanism of boric-acid-ester bonding agent in AP/RDX/Al/HTPB propellant[J]. Journal of Propulsion Technology, 2010, 31(4): 448-451. |
| [11] |
白国强, 李晓峰, 陈洛亮. 硼酸酯类键合剂研究进展[J].
化学推进剂与高分子材料, 2006, 4(6): 17-19. BAI Guo-qiang, LI Xiao-feng, CHEN Luo-liang. Research progress of borate ester bonding agents[J]. Chemcal Propellants & Polymeric Materials, 2006, 4(6): 17-19. |
| [12] |
Horst J H, Geertman R M, Van Der Heijden, et al. The influence of a solvent on the crystal morphology of RDX[J].
J Cryst Growth, 1999, 198: 773-779. |
| [13] |
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03, Revision B.02, Gaussian, Inc.[J].
Pittsburgh, PA, 2003 |
| [14] |
Rappe A K, Goddard W A. Charge equilibration for molecular dynamics simulations[J].
J Phys Chem, 1991, 95: 3358 DOI:10.1021/j100161a070 |