



2. 陕西师范大学, 陕西 西安 710062
2. Shaanxi Normal University, Xi′an 710062, China
全氮材料具有能量高、无污染的优点, 是潜在的新型高能量密度材料[1-5]。生成焓是评价全氮材料爆轰性能的关键参数。对于稳定化合物, 生成焓可由实验方法测得, 但全氮材料尚难以获得样品, 导致生成焓无法实测。因此, 采用理论计算方法成为了获取全氮材料生成焓的主要手段。然而, 如何准确可靠地计算全氮材料的生成焓仍然是一个难点。
目前, 生成焓的计算方法主要有几下几种: (1) 基团加和法、半经验分子轨道法能直接而快速地给出生成焓, 但该类方法强烈依赖于参数的准确性, 可靠性不强, 只能用于生成焓的估算; (2) 从头算方法特别是高水平理论方法如QCISD(T), CCSD(T)等能够准确地预测生成焓, 但需要巨大的计算资源, 只能用于小分子; (3) 以Gn及CBS为代表的混合方法具有很高的计算精度, 但同样需要较大的计算资源, 通常只适用于小分子体系; (4) 密度泛函理论能够合理地预测生成焓, 且只需较少的计算资源, 因此成为当前广泛使用的热门方法, 但计算结果强烈依赖于所使用的密度泛函。
本研究基于原子化反应, 采用3类9种密度泛函分别对52种多氮化合物的气相标准生成焓进行了计算, 通过比较计算值与实验值, 筛选出计算精度最高的密度泛函, 为全氮材料生成焓预测提供一种合理的方法。
2 计算方法 2.1 数据采集采集多氮化合物的生成焓数据, 包括唑、嗪、叠氮、酯、烷、硝胺、苯胺、硝基胺、硝基苯、腈等52个分子, 气相生成焓实验数据取自NIST数据库, 见表 1。
| 表 1 52个多氮化合物标准生成焓实验值与计算值偏差 Tab.1 Experiment values and deviations of standard enthalpies of formation for 52 nitrogen-rich compounds |
采用密度泛函理论B3LYP/6-31G(d)[6-7]方法对52个多氮化合物进行几何构型优化, 经振动频率计算确认所得构型为能量最优。然后, 分别采用3类9种不同的密度泛函方法进行生成焓计算, 包括(1) 单杂化密度泛函B3PW91[8]、B3P86[9]、B3LYP[6-7]、X3LYP[10]和O3LYP[11]; (2) meta杂化密度泛函M052X、M062X和M06HF[12]; (3) 双杂化密度泛函B2PLYP[13]。借助原子化反应计算生成焓, 以气相分子CaHbNcOd为例, 在标准条件下, 计算途径如图 1所示。

|
图 1 基于原子化反应计算生成焓示意图 Fig.1 Schematic of atomization reaction route to calculate the enthalpy of formation |
由图 1中的热力学循环可知, CaHbNcOd的气相生成焓ΔfH(CaHbNcOd)可通过下式求解:
| $ \begin{array}{l} {\Delta _{\rm{f}}}H({C_a}{H_b}{N_c}{O_d}) = a{\Delta _{\rm{f}}}H\left( C \right) + b{\Delta _{\rm{f}}}H\left( H \right) + c{\Delta _{\rm{f}}}H\left( N \right) + \\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;d{\Delta _{\rm{f}}}H\left( O \right)-\Delta {H_{{\rm{atomization}}}} \end{array} $ | (1) |
式中, ΔfH(C), ΔfH(H), ΔfH(N), ΔfH(O)为原子C、H、N和O的实验气相生成焓[14], kJ·mol-1; ΔHatomization为原子化反应的标准反应焓, kJ·mol-1, 通过振动频率分析可获得反应物与产物的焓值, 再经由下式计算得到:
| $ \begin{array}{l} \Delta {H_{{\rm{atomization}}}} = aH\left( C \right) + bH\left( H \right) + cH\left( N \right) + \\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;dH\left( O \right)-H({C_a}{H_b}{N_c}{O_d}) \end{array} $ | (2) |
为了更直观地体现各密度泛函的优劣, 在焓值计算中没有使用校正因子, 并统一使用Dunning基组cc-PVTZ[15-16]进行计算。所有计算均由Gaussian[17]软件完成。
2.3 误差分析采用平均偏差(MSD)和平均绝对偏差(MAD)评价气相生成焓计算值与实验值的偏离程度, 其定义分别如下:
| $ MSD = \frac{1}{n}\sum\limits_{i = 1}^n {{e_i}} $ | (3) |
| $ MAD = \frac{1}{n}\sum\limits_{i = 1}^n {|{e_i}|} $ | (4) |
式中, ei是计算值与实验值的偏差, n为分子个数, 即52。
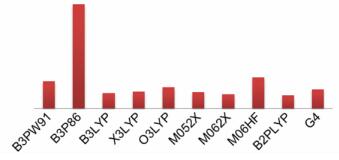
3 结果与讨论 3.1 密度泛函筛选基于原子化反应, 采用不同密度泛函计算了52个多氮化合物的气相标准生成焓, 计算偏差示于表 1。为了更客观地评价不同密度泛函的优劣, 也采用组合方法G4[18]计算了生成焓。将计算偏差按照化合物顺序作图可得图 2, 由图 2可知, B3P86与M06HF两种泛函显著偏离了其他泛函, 计算偏差最大, 尤其是B3P86泛函, 每个化合物的计算偏差均为负值, 说明计算结果存在系统误差。除了B3P86与M06HF, 其他方法的计算偏差既有正值也有负值, 随机误差可以相互抵消。

|
图 2 不同密度泛函计算的标准生成焓偏差图 Fig.2 Deviations of standard enthalpies of formation for different functionals as a function of compound number |
为了更准确地评价不同密度泛函的计算结果, 计算了52个多氮化合物气相生成焓预测值的平均偏差与平均绝对偏差。如表 2所示, M052X、M062X与B2PLYP三个泛函的平均偏差较小, 均小于15 kJ·mol-1, 说明与实验值吻合较好。其中, 以M052X泛函计算最精确。相比之下, B3P86泛函计算结果最差。若进一步区分正、负偏差对计算结果的影响, 从平均绝对偏差上看, 计算精度最高的前三种泛函分别为B2PLYP、M062X和B3LYP, 并且平均绝对偏差均小于G4方法。尽管G4方法在计算小分子原子化能时可以达到化学精度, 但并不适用大于10个原子的多氮化合物生成焓计算。因此, 最终选择双杂化泛函B2PLYP预测全氮材料的气相标准生成焓。
| 表 2 不同密度泛函计算气相生成焓的平均偏差和平均绝对偏差 Tab.2 Mean singed deviations and mean absolute deviations of standard enthalpies of formation for different functionals |
笼型全氮因具有较大环张力而备受瞩目。采用B2PLYP泛函对5种笼型全氮分子的气相标准生成焓进行了计算, 结果示于表 3。由表 3可知, 笼型全氮具有较高的正生成焓, 且随着氮原子数的增加而增加。将B2PLYP的预测结果与其他文献报道的结果进行了比较, 包括(1) 瑞典国防研究院FOI计算结果[19]; (2) 英国QinetiQ公司计算结果[20]。如图 3所示, 随着氮原子数的增加, B2PLYP计算结果的增长趋势与FOI计算结果类似, 但生成焓数据整体偏小。相比之下, QinetiQ计算结果的增长趋势较为平缓。
| 表 3 笼型全氮分子的气相标准生成焓 Tab.3 Standard enthalpies of formation for representative all-nitrogen molecules with cage type |

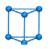

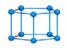


|
图 3 生成焓预测值与笼型全氮原子数关系图 Fig.3 Predicted enthalpies of formation for all-nitrogen molecules with cage type as a function of nitrogen atom number |
通常, 在预估含能材料爆轰性能时更关注的是固相生成焓。考虑到气相生成焓的实验数据比固相生成焓多, 为了准确评价不同密度泛函的优劣, 需采集尽可能多的数据, 因此本研究只计算了气相生成焓。至于全氮材料的固相生成焓, 可根据本系列论文“Ⅰ.晶体密度预测”[21]中预测的晶体结构进行晶格能计算, 从而获得固相生成焓。
4 结论(1) 基于原子化反应, 通过3类9种密度泛函分别对52种多氮化合物的气相标准生成焓进行了计算, 双杂化泛函B2PLYP的计算精度最高, 平均绝对偏差为30.1 kJ·mol-1
(2) 采用B2PLYP泛函预测了5种笼型全氮分子N4(Td), N6(D3h), N8(Oh), N10(D5h), N12(D6h)的气相生成焓分别为756.4, 1338.2, 1878.5, 2144.3, 2787.0 kJ·mol-1, 随着氮原子数目的增加, 生成焓逐渐增加。
| [1] | Eremets M I, Gavriliuk A G, Trojan I A, et al. Single-bonded cubic form of nitrogen[J]. Nature Materials, 2004, 3(8): 558-563. DOI:10.1038/nmat1146 |
| [2] | Samartzis P C, Wodtke A M. All-nitrogen chemistry: how far are we from N60?[J]. International Reviews in Physical Chemistry, 2006, 25(4): 527-552. DOI:10.1080/01442350600879319 |
| [3] | Hirshberg B, Gerber R B, Krylov A I. Calculations predict a stable molecular crystal of N8[J]. Nature Chemistry, 2014, 6(1): 52-56. |
| [4] |
李玉川, 庞思平. 全氮型超高能含能材料研究进展[J].
火炸药学报, 2012, 35(1): 1-8. LI Yu-chuan, PANG Si-ping. Progress of all-nitrogen ultrahigh-energetic material[J]. Chinese Journal of Explosive & Propellants, 2012, 35(1): 1-8. |
| [5] |
张光全, 董海山. 氮簇合物--潜在的高能量密度材料候选物[J].
含能材料, 2004, 12(增刊): 105-113. ZHANG Guang-quan, DONG Hai-shan. Nitrogen clusters-potential candidates as high-energy density materials[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2004, 12(supple): 105-113. |
| [6] | Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Physical Review B, 1988, 37(2): 785-789. DOI:10.1103/PhysRevB.37.785 |
| [7] | Becke A D. Density-functional thermochemistry . Ⅲ. The role of exact exchange[J]. Journal of Chemical Physics, 1993, 98(7): 5648-5652. DOI:10.1063/1.464913 |
| [8] | Perdew J P, Wang Y. Accurate and simple analytic representation of the electron gas correlation energy[J]. Physical Review B, 1992, 45(23): 13244-13249. DOI:10.1103/PhysRevB.45.13244 |
| [9] | Perdew J P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J]. Physical Review B, 1986, 33(12): 8822-8824. DOI:10.1103/PhysRevB.33.8822 |
| [10] | Xu X, Goddard Ⅲ W A. The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties[J]. Proceedings of the National Academy of Science, 2004, 101(9): 2673-2677. DOI:10.1073/pnas.0308730100 |
| [11] | Cohen A J, Handy N C. Dynamic correlation[J]. Molecular Physics, 2001, 99(7): 607-615. DOI:10.1080/00268970010023435 |
| [12] | Zhao Y, Truhlar D G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals[J]. Theoretical Chemistry Accounts, 2008, 120(1-3): 215-241. DOI:10.1007/s00214-007-0310-x |
| [13] | Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation[J]. The Journal of Chemical Physics, 2006, 124: 034108 DOI:10.1063/1.2148954 |
| [14] | http://webbook.nist.gov/chemistry/2016.01.10. |
| [15] | Head-Gordon M, Pople J A, Frisch M J. MP2 energy evaluation by direct methods[J]. Chemical Physics Letters, 1988, 153(6): 503-506. DOI:10.1016/0009-2614(88)85250-3 |
| [16] | Kendall R A, Dunning T H, Harrison R J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions[J]. The Journal of Chemical Physics, 1992, 96(9): 6796-6806. DOI:10.1063/1.462569 |
| [17] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09[CP], Gaussian, Inc., Wallingford CT, 2009. |
| [18] | Curtiss L A, Redfern P C, Raghavachari K. Gaussian-4 theory[J]. Journal of Chemical Physics, 2007, 126: 084108 DOI:10.1063/1.2436888 |
| [19] | Östmark H. High energy density materials (HEDM): overview, theory and synthetic efforts at FOI[C]//New Trends in Research o f Energetic Materials, Czech Republic, 2006: 231-250. |
| [20] | Haskins P J, Fellows J, Cook M D, et al. Molecular level studies of polynitrogen explosives[C]//12th International Detonation Symposium, California, 2002. |
| [21] |
刘英哲, 来蔚鹏, 尉涛, 等. 全氮材料基础性能理论研究:Ⅰ.晶体密度预测[J].
含能材料, 2017, 25(2): 100-105. LIU Ying-zhe, LAI Wei-peng, WEI Tao, et al. Theoretical investigations on fundamental properties of all-Nitrogen materials: Ⅰ.Predication of crystal densities[J]. Chinese Journal of Energetic Materials(Hanneng Cailiao), 2017, 25(2): 100-105. DOI:10.11943/j.issn.1006-9941.2017.02.002 |
The enthalpies of formation of 52 nitrogen-rich compounds were calculated by different density functionals.